Pheochromocytoma is a rare but life-threatening condition that has varied clinical presentations particularly hypertension, headache, palpitation, and sweating.1,2 Patients with suggestive clinical features are frequently tested for pheochromocytoma. The medical interest in this tumor has increased with the improved availability of diagnostic laboratory tools particularly plasma or urinary fractionated metanephrines (metanephrine and normetanephrine), and other neuroendocrine markers particularly chromogranin A.3,4 The wide universal availability of different imaging facilities, both anatomical and functional, has also improved the detection of pheochromocytoma.5
The growing awareness for implementing different protocols and guidelines that consider pheochromocytoma in the work-up and differential diagnosis has improved diagnosis of the disorder.6 Accurate diagnosis is important because if the tumor is detected early laparoscopic or surgical treatment is usually curative before other changes or complications take place.
Catecholamine-producing tumors are neuroendocrine tumors that affect the chromaffin cells of adrenal medulla and postganglionic fibers of the sympathetic nervous system.1,2 These tumors are characterized by the synthesis, storage, release, and secretion of catecholamines and their metabolites. They include pheochromocytomas in the adrenal medulla and paragangliomas in the extra-adrenal sympathetic ganglions usually below the diaphragm in the retroperitoneum or organ of Zuckerkandl and various sites including the head, neck, thorax, and abdomen. However, although the majority of these tumors are benign and adrenal, investigation workup should consider their tendency for being multiple, malignant, and familial with genetic pathogenesis.7
Epinephrine (and its metabolite metanephrine) is the catecholamine that is produced exclusively by the adrenal medulla and adrenal pheochromocytoma. On the other hand, norepinephrine (and its metabolite normetanephrine) is the catecholamine produced by the adrenal medulla and adrenal pheochromocytoma as well as by the extra-adrenal pheochromocytoma and paraganglioma, which can also produce dopamine and its metabolite methoxytyramine. However, this is rare.8 The biochemical synthesis of the different catecholamines and their metabolites is shown in Figure 1.
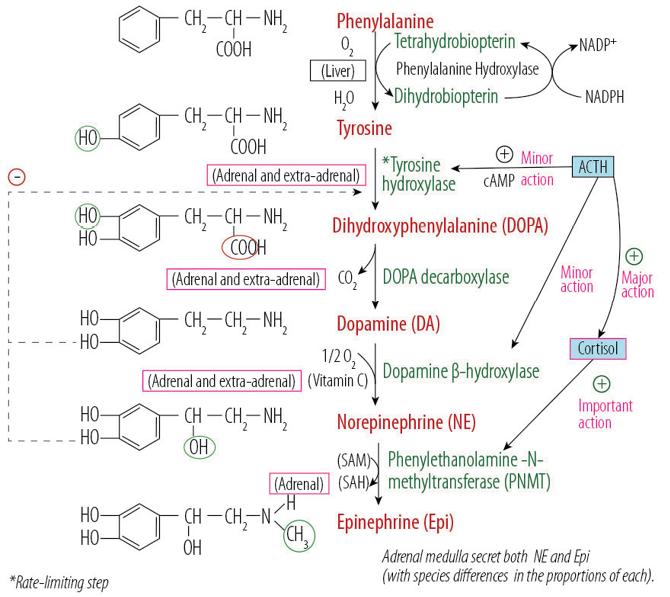
Figure 1: Adrenal medulla catecholamines biosynthesis showing the required enzymes, coenzymes, and the influencing hormonal effect. In extra-adrenal sympathetic fibers the biosynthesis stops at the production of norepinephrine while in the adrenal medulla the end product of catecholamines synthesis is epinephrine.
Pheochromocytomas can affect individuals of all ages. They are common in people aged between 40 and 50 years, and relatively more common among females. Adrenal pheochromocytomas constitute nearly 85% of cases of pheochromocytomas, with 15% being extra-adrenal paragangliomas that affect the sympathetic ganglions anywhere from the base of the brain to the urinary bladder.1,2 Laboratory diagnosis is usually achieved by measuring the catecholamines, or more importantly their metabolites in plasma or urine. Although different markers, including catecholamines and vanillylmandelic acid, are utilized as diagnostic tests, total and fractionated metanephrines in plasma or urine provide the best valid laboratory test for excluding or confirming pheochromocytoma.2,9 In addition, chromogranin A is another reliable marker for neuroendocrine tumors, including pheochromocytoma and is increasingly used in the diagnostic workup and follow-up of patients with these tumors.4 Following diagnosis, removal of the adrenal gland or the tumor is done by open surgery or by the laparoscopic technique, which is considered the ‘gold standard’ treatment choice since 1992.10-12
Case Report
A 50-year-old female presented to the Outpatient Clinic, Royal Hospital, Oman, with abdominal pain in the right upper quadrant of the abdomen and backache that had lasted 10 days. The patient’s history revealed that she had similar abdominal pain two months prior. The pain was a “dull ache” and somewhat associated with a headache. The pain was relieved with a mild analgesic drug (acetaminophen). She had no difficulty in breathing and no cough. No changes in bowel habits, nausea, or vomiting were reported. The patient did not give a history of hypertension, diabetes, ischaemic heart disease, or chronic obstructive airway disease. She had three lower segment cesarean sections; the last one dated 18 years ago. She was not on any medications other than the occasional analgesic drug for headaches. She was not a smoker or a consumer of alcohol. Menses had become infrequent during the last two years.
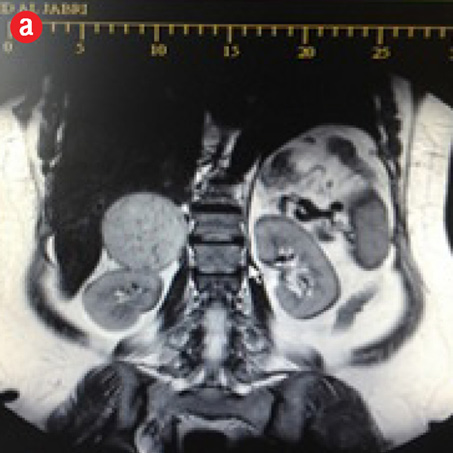
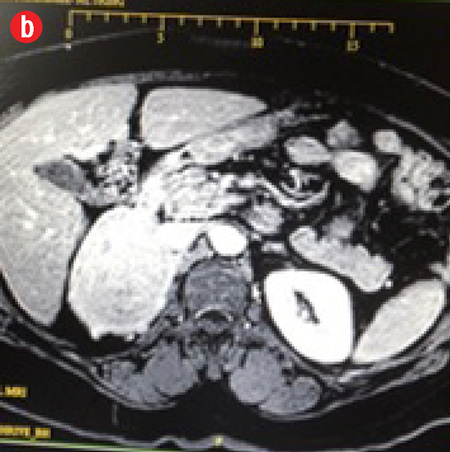
The patient was a middle aged, obese, female, with a pulse of 84 breaths/min, and a blood pressure (BP) of 122/78mmHg. Physical examination revealed no pallor, jaundice, or lymphadenopathy. Abdominal examination showed a well-healed Pfannenstiel scar, a soft abdomen, no tenderness, no palpable masses, no organomegaly, and no ascites. Her bowel sounds were normal. She had daily recordings of her BP for one month, which were always normal. An ultrasonography of the abdomen showed a right adrenal mass. She then had computed tomography (CT) scan of the abdomen and later magnetic resonance imaging (MRI) of her adrenal glands. The MRI confirmed a right adrenal mass (7.3 × 6.0 × 5.5 cm) that was hypervascular with areas of internal hemorrhage. These features were consistent with a pheochromocytoma [Figure 2]. The mass appeared to compress the inferior vena cava, especially at the site of drainage of the right renal vein, which was also slightly compressed by the mass. Both veins were patent with no evidence of invasion or thrombosis. The mass was located just above the course of the right renal artery, which was touching it, with no evidence of right renal artery invasion and no encasement. The right kidney was of normal dimensions and looked normal. The left adrenal looked unremarkable, with a normal left kidney.
|
|
|
|
|
Figure 2: Magnetic resonance imaging confirming a right adrenal mass (7.3 × 6.0 ×5.5 cm) that was hypervascular with areas of internal hemorrhage. These features are consistent with a pheochromocytoma. (a) Pre-contrast view. (b) Post-contrast view. |
All laboratory data are shown in Table 1. The patient had laboratory results within the references ranges for core blood laboratory tests including electrolytes, renal, liver, bone, glucose, glycated hemoglobin (HbA1c), and thyroid profiles. Endocrine tests revealed normal levels of aldosterone and renin and low-normal levels of serum cortisol. Plasma catecholamines (dopamine, norepinephrine, and epinephrine) levels were within the reference ranges. Plasma metanephrine levels were normal at the time of consultation and prior to surgery. Plasma normetanephrine levels were markedly raised (10-fold) at the time of consultation and prior to surgery (20-fold). Additionally, plasma chromogranin A was markedly raised (16-fold) at time of consultation and prior to surgery (15-fold). The laboratory profile was not compatible with adrenal pheochromocytoma but was with extra-adrenal pheochromocytoma. One week after surgery, normetanephrine and chromogranin A levels were normal.
Table 1: Plasma levels of catecholamines (and their metabolites), renin, aldosterone, cortisol, and chromogranin A compared to the recommended adult’s reference ranges in the preoperative week. One- week post-operative levels are provided for plasma metanephrine, normetanephrine and 24-hour urine cortisol.
|
Dopamine (pmol/L) |
<1.0 |
- |
≤1.0 |
|
Norepinephrine (pmol/L) |
3.2 |
- |
≤4.0 |
|
Epinephrine (pmol/L) |
<1.0 |
- |
≤1.0 |
|
Metanephrine (nmol/L) |
0.1 |
<0.1 |
≤0.5 |
|
Normetanephrine (nmol/L) |
20.1 |
0.5 |
≤1.0 |
|
Chromogranin A (IU/L) |
323.0 |
26.0 |
≤22.0 |
|
Aldosterone (pmol/L) |
130.0 |
- |
70–440 (recumbent), 110–860 (upright) |
|
Renin (mIU/L) |
5.3 |
- |
supine 4–40, upright 6–60 |
|
10 am cortisol (nmol/L) |
255.0 |
- |
200–550 |
*Postoperative specimens were taken one week following adrenalectomy.
The patient was admitted one day prior to surgery for preparation and assessment. All precautions were taken and preparations for managing the preoperative hypertensive crisis.
Laparoscopic right adrenalectomy was performed. During surgery, while dissecting near the main adrenal vein, her BP rose to 240/120mmHg. After clipping and division of the main adrenal vein and accessory adrenal vein then it came down rapidly to normal levels. There was some blood loss that necessitated two units of packed red blood cells (RBC) transfusion. The adrenal tumor was excised and retrieved in toto [Figure 3]. The patient did well and remained normotensive in the postoperative period. She was discharged after eight days without complaint and with normal neuroendocrine markers. She refused genetic testing.
Figure 3: Gross macroscopic appearance of the resected adrenal gland including the pheochromocytoma tumor. (a) and (b) Laparoscopic views of the right adrenal mass. (c) Gross macroscopic appearance of the right adrenal mass specimen (anterior surface).
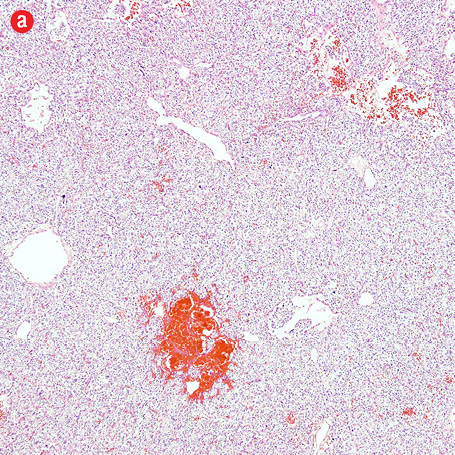
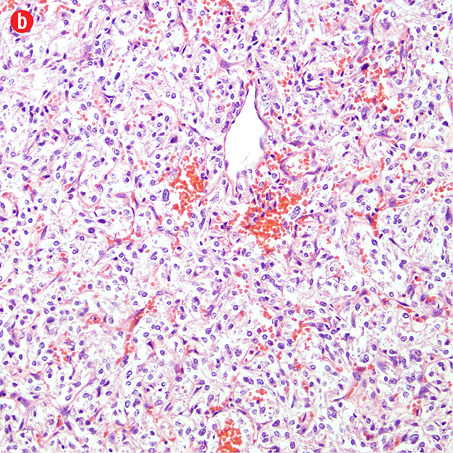
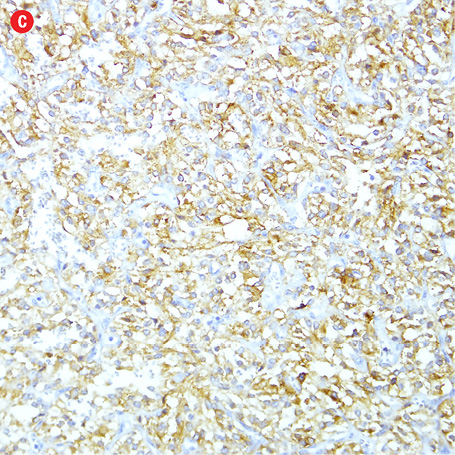
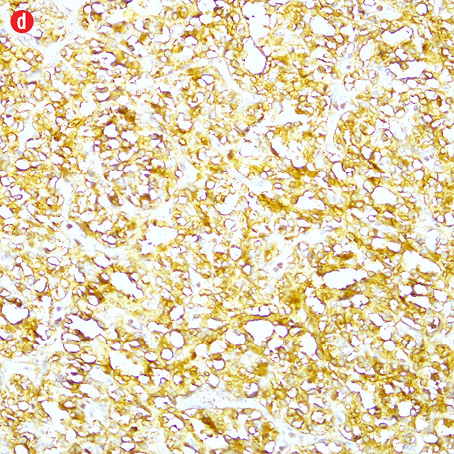
Gross examination of the right adrenal gland showed a nodular mass with the measurements of 9.0 × 6.5 × 4.0 cm and weighed 159g. The capsule was ruptured at places, and the cut section appeared brown and hemorrhagic. The microscopy sections showed a neoplasm comprising of intermediate to large cells, arranged in an alveolar pattern. The tumor cells were surrounded by a thin capillary network (Zellballen appearance). Occasional cells had large bizarre nuclei. There were areas of hemorrhage, cystic changes, and aggregates of hemosiderophages. Mitotic figures or necrotic foci were not present. There was no evidence of vascular invasion. An uninvolved adrenal gland was seen at the periphery. The tumor cells were focally infiltrating through the capsule [Figure 4]. Immunohistochemistry of the tumor cells were positive for chromogranin, synaptophysin, and S-100 protein. They were negative for AE1/AE3, inhibin, HMB-45, and cytokeratin. Ki-67 protein showed a proliferative index of less than 5%.
|
|
|
|
|
|
|
|
|
Figure 4: Histopathology of the resected adrenal gland (including the tumor). Histopathology showed: (a) adrenal pheochromocytoma. An anastomosing trabecular pattern of tumor cells separated by thin vascular channels. Occasional bizarre cells were present, magnification=10×. (b) Foci of lipid-laden cells, magnification=20×. (c) Spindle cells in a hyalinised stroma, magnification=20×. (d) Chromogranin strong positivity in tumor cells, magnification=20×. |
A stepwise analysis of pheochromocytoma genes, MAX, SDHA, SDHAF2, SDHB, SDHC, SDHD, VHL, and PRKAR1A (HGMD® Professional 2014.4) was performed at Centogene Laboratory (Centogene AG, Schillingallee, Germany). No pathogenic mutations were detected. However, no deletion/duplication analysis to test for mutations due to large deletions/duplications was conducted. The SDHB and VHL genes were analysed by the polymerase chain reaction (PCR) and sequencing of both DNA strands of the entire coding region and the highly conserved exon-intron splice junctions was performed.
The MAX, PRKAR1A, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, and VHL genes were analysed by PCR and next-generation sequencing of both DNA strands of the entire coding region and the highly conserved exon-intron splice junctions was performed. A PCR-based amplicon library capture was utilized.
Discussion
The diagnosis of pheochromocytoma and paraganglioma usually requires biochemical confirmation of excessive catecholamines production and imaging investigation for detecting the tumor.1-4 Awareness and clinical suspicion of these neuroendocrine tumors in patients with classical or non-classical presentation is a key point in planning workup investigations. Increasing availability of fractionated metanephrines assays particularly in plasma, which has almost replaced 24-hour urine, in many laboratories using universal immunoassay equipment has improved the diagnostic efficiency.6,9
In our laboratory, we have established plasma fractionated metanephrines (metanephrine and normetanephrine) measurement using the enzyme-linked immunosorbent assay (ELISA) technology for the last three years in compliance with the recent guidelines for diagnosing pheochromocytoma.9 Since then the requests for plasma (instead of timed urine) metanephrines has increased by four- to five-fold. The availability of chromogranin A as another biochemical neuroendocrine marker has added to the diagnostic tools for diagnosing neuroendocrine tumors including catecholamine secreting tumors.
Further improvement in the diagnosis of these disorders is expected with the current trend towards automating these tests that is expected to be implemented in practice shortly. Workup search for important risk problems has to consider different underlying metabolic disorders in patients with various clinical scenarios that may be presented with bizarre manifestations including cardiovascular diseases.13,14
We reported a rare case of right adrenal pheochromocytoma in a 50-year-old Omani woman with unusual clinical and biochemical expression. There were at least three unusual findings in this patient. Firstly, the lack of hypertension, palpitation, or sweating, which are classical for pheochromocytoma, secondly, the normal level of plasma catecholamines, and thirdly, the normal level of plasma metanephrine despite the high levels of plasma normetanephrine and chromogranin A. The clinical, imaging, and histopathology findings confirmed adrenal pheochromocytoma; however, the biochemical endocrine profile was consistent with extra-adrenal pheochromocytoma. A literature search in Pubmed in August 2015 suggests that our patient with such bizarre biochemical and clinical presentation is the first reported case in Oman.
The lack of classical features of pheochromocytoma in our case can be explained by the normal level of plasma catecholamines, which excludes excess catecholamines production reaching the circulation and hence lack of resultant supra-physiological response to these neurotransmitters. Although plasma or urine catecholamines are usually raised in pheochromocytoma and paraganglioma, there are a few reported cases with normal level. In all these tumors, whether catecholamines are raised or normal, there are raised levels of the O-methylated metabolites of norepinephrine and epinephrine namely normetanephrine and metanephrine, respectively, that are produced by the enzyme catechol-O-methyltransferase (COMT) on their corresponding parent hormone.
Hence, measurement of these catecholamines’ metabolites has been recommended to replace catecholamines measurement for diagnosing catecholamine-secreting tumors, both adrenal pheochromocytomas (associated with increased metanephrine and normetanephrine) and extra-adrenal paragangliomas (associated with increased normetanephrine only).
In our case, despite our expectation that the patient would have raised epinephrine and metanephrine due to her large adrenal pheochromocytoma, she had normal plasma level of this catecholamine and its metabolite. However, the patient had only markedly raised normetanephrine, a biochemical pattern characteristic for extra-adrenal pheochromocytoma or paraganglioma but not adrenal pheochromocytoma.
Two pathophysiological mechanisms can explain this unusual observation in this biochemical expression of the tumor. The normal level of plasma catecholamines and high level of the O-methylated metabolite, normetanephrine, indicates that there is high level of the catecholamine norepinephrine produced by the large adrenal tumor. However, the discrepancy between catecholamine level and its metabolites indicates that the catecholamine itself is metabolized within the tumor and converted to its O-methylated metabolite, normetanephrine, before its secretion into the circulation. Thereafter, it is metabolized mainly in liver and kidney. Hence, no symptoms of catecholamine excess were noted in this patient, apart from the abdominal pain due to the physical burden of the large tumor in the abdomen. Patients with epinephrine secreting pheochromocytoma usually show more frequent symptoms than norepinephrine secreting pheochromocytoma.15,16
Eisenhofer et al,17 in their study using western blot analysis, enzyme assay, and immunohistochemistry confirmed the presence in pheochromocytomas of the COMT enzyme that is responsible for the conversion of catecholamines (norepinephrine and epinephrine) to their metabolites (normetanephrine and metanephrine). This intra-tumor COMT is mostly in membrane bound form and less frequently in soluble form. Other researchers,18-20 have reported comparable observations in patients with pheochromocytoma without catecholamines secretion, or with episodic secretion. This intra-neoplasm metabolism of catecholamines to free metanephrines would explain the higher sensitivity and advantage of measuring the free metanephrines compared to the parent catecholamines and other metabolites for the diagnosis of these tumors.2,9
In addition to normal plasma catecholamines levels, the patient had normal metanephrine with markedly raised normetanephrine (20-fold). This biochemical pattern is unexpected for adrenal pheochromocytoma; however, it is compatible with extra-adrenal pheochromocytoma and paraganglioma. From a physiological view point, adrenal medulla and adrenal pheochromocytoma produce epinephrine (which can be metabolized to metanephrine) that are exclusively produced and secreted by these chromaffin cells. On the other hand, extra-adrenal sympathetic fibres and paragangliomas cannot produce epinephrine but catecholamine production ends by norepinephrine (which can be metabolized to normetanephrine), with both can also be produced by the adrenal medulla and pheochromocytoma but in lesser amounts than epinephrine and metanephrine. The physiological basis for the unique production of epinephrine by the adrenal medulla is that in the biosynthesis of catecholamines, the conversion of norepinephrine to epinephrine is catalysed by the enzyme phenylethanolamine N-methyl transferase (PNMT); a process that occurs only in the adrenal chromaffin cells. The expression of this PNMT is controlled by glucocorticoid-receptor mediated mechanisms acting in collaboration with many transcription factors.21-25 The local availability of steroids in the anatomical surroundings of the adrenal cortex allow the adrenal medulla chromaffin cells to be exposed to very high concentration of glucocorticoids (more than 100-fold its concentration in the systemic circulation) that will induce PNMT synthesis and hence epinephrine production. Accordingly, adrenal pheochromocytomas often produce epinephrine in addition to norepinephrine. In contrast, extra-adrenal sympathetic fibres and paragangliomas typically lack PNMT, so catecholamines production stops at norepineprine with no conversion to epinephrine and extra-adrenal pheochromocytomas can produce only norepinephrine and not epinephrine.26
In patients with large pheochromocytoma, as in our patient with her adrenal tumor, the large size of the tumor can cause expansion of the tumor resulting in dissociation of the adrenal medulla from the adrenal cortex and the adrenal medulla will lose its vascular contact and paracrine control from the adrenal cortex. Hence, in large adrenal medulla tumors, the aforementioned influence of glucocorticoids on PNMT induction will be lost.
In our patient, with her right adrenal pheochromocytoma and due to its large size, the tumor produced only norepinephrine that is metabolized by COMT within the tumor itself into normetanephrine. Therefore, plasma norepinephrine was within normal range with markedly raised normetanephrine. Meanwhile, both epinephrine and metanephrine were within the normal range because of failure of formation of epinephrine and its metabolite, metanephrine due to the large size of the adrenal tumor and its impact on glucocoriticoid driven PNMT action on conversion of epinephrine to norepinephrine. Hence, the only marker of catecholamine excess in our patient was raised plasma metanephrine and chromogranin A.
Chromogranin A is a member of the granin family of neuroendocrine secretors, which is secreted in equimolar basis with all catecholamines from the secretory and storage granules of chromaffin cells. It is not metabolized in the adrenal medulla, adrenal tumor, or the body. Its concentration in the circulation is a good reflector of neurocrine activity both at the physiological and supraphysiological level and in patients with neuroendocrine tumors including pheochromocytoma, carcinoid, gastrinoma, primary hyperparathyroidism, and other tumors its measurement represent a reliable marker for the diagnosis and management of patients with these tumors.27
No pathogenic mutation in the pheochromocytoma genes MAX, SDHA, SDHAF2, SDHB, SDHC, SDHD, VHL, and PRKAR1A (Human Gene Mutation Database; HGMD® Professional 2014.4) were detected in our patient. However, no deletion/duplication analysis to test for mutations due to large deletions/duplications was conducted. The reasons for not identifying a mutation may be due to the location of the mutation in an intron or a regulatory element of the examined genes, which are not included in routine genetic diagnostics, the location of the mutation in another gene that is still not reported as associated with the phenotype, or the phenotype is not driven by a genetic mutation, and so the patient has no underlying genetic abnormality.
Genetic testing is usually indicated in patients with a positive family history, young age (<45 years) at the time of manifestation, the presence of multiple tumors, extra-adrenal location, malignant neoplasm, and previous head and neck paraganglioma as well as the presence of distinct clinical syndromes.28 These syndromes are usually caused by one of 12 susceptibility genes namely VHL (von Hippel-Lindau disease; caused by germline mutations in the VHL tumor suppressor gene), RET (in multiple endocrine neoplasia type 2; MEN 2), SDHD, SDHAF2, SDHC, and SDHB (in paraganglioma syndrome types 1–4), NF1 gene (in type 1 neurofibromatosis; NF), SDHA, TEMEM127, MAX, EPAS1 and H-RAS genes (in familial pheochromocytoma syndromes).7,29,30 KIF1B, EGLN1, IDH1, and FH genes have been reported to be associated with hereditary paraganglioma-pheochromocytoma, but their clinical significance is still unclear.31
Conclusion
We have reported a rare case of large right adrenal pheochromocytoma with unusual clinical and biochemical expression in a middle-aged Omani woman, which the patient completely recovered from after tumor resection. Clinical, imaging, and histopathology findings confirmed adrenal pheochromocytoma. However, her biochemical endocrine profile was consistent with extra-adrenal pheochromocytoma and no pathogenic mutation in pheochromocytoma genes were detected.
Disclosure
The molecular testing of this patient was sponsored by Health 4 All LLC, Oman (www.health-4all-oman.com). The authors declared no conflicts of interest.
references
- Manger WM, Gifford RW. Pheochromocytoma. J Clin Hypertens (Greenwich) 2002 Jan-Feb;4(1):62-72.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005 Aug;366(9486):665-675.
- Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 2002 Mar;287(11):1427-1434.
- Grossrubatscher E, Dalino P, Vignati F, Gambacorta M, Pugliese R, Boniardi M, et al. The role of chromogranin A in the management of patients with phaeochromocytoma. Clin Endocrinol (Oxf) 2006 Sep;65(3):287-293.
- Havekes B, King K, Lai EW, Romijn JA, Corssmit EP, Pacak K. New imaging approaches to phaeochromocytomas and paragangliomas. Clin Endocrinol (Oxf) 2010 Feb; 72(2):137-145.
- Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB, et al; International Symposium on Pheochromocytoma. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab 2007 Feb;3(2):92-102.
- Rana HQ, Rainville IR, Vaidya A. Genetic testing in the clinical care of patients with pheochromocytoma and paraganglioma. Curr Opin Endocrinol Diabetes Obes 2014 Jun;21(3):166-176.
- Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 2012 Jul;48(11):1739-1749.
- Unger N, Deutschbein T, Walz MK, Mann K, Petersenn S. The value of immunoassays for metanephrines in the biochemical diagnosis of pheochromocytomas. Horm Metab Res 2009 Sep;41(9):676-679.
- Gumbs AA, Gagner M. Laparoscopic adrenalectomy. Best Pract Res Clin Endocrinol Metab 2006 Sep;20(3):483-499.
- Conzo G, Musella M, Corcione F, De Palma M, Ferraro F, Palazzo A, et al. Laparoscopic adrenalectomy, a safe procedure for pheochromocytoma. A retrospective review of clinical series. Int J Surg 2013;11(2):152-156.
- Assalia A, Gagner M. Laparoscopic adrenalectomy. Br J Surg 2004 Oct;91(10):1259-1274.
- Ba Arah BT. A case of von Hippel-Lindau disease with exudative maculopathy. Oman J Ophthalmol 2009 May;2(2):82-85.
- Pieris RR, Al-Sabti HA, Al-Abri QS, Rizvi SG. Prevalence pattern of risk factors for coronary artery disease among patients presenting for coronary artery bypass grafting in Oman. Oman Med J 2014 May;29(3):203-207.
- Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 2001 May;86(5):1999-2008.
- Ito Y, Fujimoto Y, Obara T. The role of epinephrine, norepinephrine, and dopamine in blood pressure disturbances in patients with pheochromocytoma. World J Surg 1992 Jul-Aug;16(4):759-764.
- Eisenhofer G, Keiser H, Friberg P, Mezey E, Huynh TT, Hiremagalur B, et al. Plasma metanephrines are markers of pheochromocytoma produced by catechol-O-methyltransferase within tumors. J Clin Endocrinol Metab 1998 Jun;83(6):2175-2185.
- Sinclair D, Shenkin A, Lorimer AR. Normal catecholamine production in a patient with a paroxysmally secreting phaeochromocytoma. Ann Clin Biochem 1991 Jul;28(Pt 4):417-419.
- Stewart MF, Reed P, Weinkove C, Moriarty KJ, Ralston AJ. Biochemical diagnosis of phaeochromocytoma: two instructive case reports. J Clin Pathol 1993 Mar;46(3):280-282.
- Shawar L, Svec F. Pheochromocytoma with elevated metanephrines as the only biochemical finding. J La State Med Soc 1996 Dec;148(12):535-538.
- Wurtman RJ, Axelrod J. Control of enzymatic synthesis of adrenaline in the adrenal medulla by adrenal cortical steroids. J Biol Chem 1966 May;241(10):2301-2305.
- Wong DL, Siddall BJ, Ebert SN, Bell RA, Her S. Phenylethanolamine N-methyltransferase gene expression: synergistic activation by Egr-1, AP-2 and the glucocorticoid receptor. Brain Res Mol Brain Res 1998 Oct;61(1-2):154-161.
- Wong DL, Siddall B, Wang W. Hormonal control of rat adrenal phenylethanolamine N-methyltransferase. Enzyme activity, the final critical pathway. Neuropsychopharmacology 1995 Nov;13(3):223-234.
- Eisenhofer G, Huynh TT, Hiroi M, Pacak K. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev Endocr Metab Disord 2001 Aug;2(3):297-311.
- Zuckerman-Levin N, Tiosano D, Eisenhofer G, Bornstein S, Hochberg Z. The importance of adrenocortical glucocorticoids for adrenomedullary and physiological response to stress: a study in isolated glucocorticoid deficiency. J Clin Endocrinol Metab 2001 Dec;86(12):5920-5924.
- Osamura RY, Kawai OY, Hori S, Suemizu H, Onoda N, Joh TH. Immunohistochemical localization of catecholamine-synthesizing enzyme in human pheochromocytomas. Endocr Pathol 1990 Jun;1(2):102-108.
- Modlin IM, Gustafsson BI, Moss SF, Pavel M, Tsolakis AV, Kidd M. Chromogranin A–biological function and clinical utility in neuro endocrine tumor disease. Ann Surg Oncol 2010 Sep;17(9):2427-2443.
- Erlic Z, Rybicki L, Peczkowska M, Golcher H, Kann PH, Brauckhoff M, et al; European-American Pheochromocytoma Study Group. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res 2009 Oct;15(20):6378-6385.
- Bausch B, Malinoc A, Maruschke L, Offergeld C, Gläsker S, Rischke HC, et al. [Genetics of pheochromocytoma]. Chirurg 2012 Jun;83(6):511-518.
- Crona J, Nordling M, Maharjan R, Granberg D, Stålberg P, Hellman P, et al. Integrative genetic characterization and phenotype correlations in pheochromocytoma and paraganglioma tumours. PLoS One 2014;9(1):e86756.
- Kirmani S, Young WF. Hereditary paraganglioma-pheochromocytoma syndromes. In: Pa-gon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K (edi-tors). Gene Reviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Updated Nov 2014.