|
Abstract
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an X-linked genetic disorder characterized by low levels of the G6PD enzyme. It is present worldwide but with more prevalence in the Middle East and the Mediterranean areas. We report a case of severe hemolysis due to G6PD deficiency manifesting as methemoglobinemia in a 70 year old Omani male never known to have any previous hemolytic episodes or previously diagnosed of G6PD deficiency.
Keywords: G6PD deficiency; Hemolysis; Methemoglobinemia.
Introduction
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common enzymatic deficiency of red blood cells in humans, affecting around 400 million people around the world.1 The condition is most prevalent in the Mediterranean and the Middle East areas. The enzyme G6PD is involved in the pentose phosphate pathway which is essential for red cell metabolism. It leads to the generation of NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) which keeps the hemoglobin (Hb) heme iron in the ferrous state.
There are number of variants of this disease with variable deficiency levels and clinical manifestations.1,2 Severe deficiency (<10% activity) manifests as chronic hemolytic anemia. The most common variant in Oman is the Mediterranean variant.2 It is well known that there are number of precipitating factors for hemolysis in this condition, including fava beans and henna. Though many patients in Oman eat fava beans without having any hemolytic consequences, some develop severe hemolytic attacks with Hb levels dropping down to as low as 5 g/dL following exposure to henna.3
Severe hemolysis due to G6PD deficiency may manifest as methemoglobinemia,4 in which the heme iron is in the oxidized ferric state rather than the ferrous state.5 This resultant hemoglobin, known as methemoglobin (met-Hb), cannot carry oxygen and the remaining oxyhemoglobin develops increased oxygen affinity resulting in impaired oxygen delivery. This results in a left shifted oxygen–hemoglobin dissociation curve and secondary tissue hypoxia. Normal people generate met-Hb but in very low levels in the range of 0.5% to 3%.6
Met-Hb has been reported to present for the first time during adulthood.7 However, to the best of our knowledge, it has not been reported to present amongst the elderly. We report a case of an elderly man who presented with methemoglobinemia as the first manifestation of severe G6PD deficiency related hemolysis.
Case Report
A 70-year-old Omani male was brought by his son to the Accident and Emergency Department (AE) with a history of jaundice and lethargy of less than 24-hours duration. Past medical history was significant for dementia for which he was on olanzepine. There was no family history of any similar problems.
His examination revealed a fully awake, deeply jaundiced man with a slightly cyanosed lips and tongue. He was confused which was attributed to his dementia. He had a heart rate of 100 bpm, blood pressure of 180/90 mmHg and a temperature of 39°C without any obvious focus of infection. His oxygen saturation on the pulse-oximeter was 70% in room air, while his arterial blood gases (ABG) showed oxygen saturation of 90% and a met-Hb of 18%.
Complete blood count showed a hemoglobin of 11 g/dL (11.5-15.5), (one year ago, Hb was 15 g/dL), white blood cells count (WBC) of 24 × 109/L (2.2-10.0) with neutrophilia at 19 × 109/L (1.0-5.0), platelets of 328 × 109/L (140-400). Absolute reticulocyte was at 68 × 109/L (20.0-150.0). Liver function tests showed aspartate aminotransferase of 59 U/L (0-40), alanine aminotransferase of 25 IU/L (0-41), albumin of 46 (35-52), alkaline phosphatase of 160 U/L (40-129) and bilirubin of 282 umol/L (0-17) with an unconjugated bilirubin of 260 umol/L. Renal function tests showed urea levels of 11.3 mmol/L (2.8-8.1), creatinine of 121 umol/L (59-104), sodium of 142 mmol/L (135-145), potassium of 4.6 mmol/L (3.5-5.1), and HCO3 of 24 mmol/L (22-29). Lactate dehydrogenase level (LDH) was 949 U/L (135-225). His coagulation screen was normal. The patient passed 400 mL of urine in the emergency department with macroscopic hematuria. Chest X-ray was normal, and blood and urine culture were later reported to have no growth.
A diagnosis of methemoglobinemia was made on the basis of the gap between the oxygen saturation on the monitor and on the ABGs and on the met-Hb% as measured by the blood gas analysis machine. A serial of ABGs showed gradual increment of the met-Hb level but because his G6PD levels were <30%, he could not be given methylene blue. He was managed with 100% oxygen, generous hydration and the broad spectrum antibiotic meropenem.
Five hours later, his Hb had dropped to 9.6 g/dL, and platelets to 279 × 109/L. Blood film (Fig. 1) showed a leukoerythroblastic picture with marked polychromasia, large number of circulating nucleated red blood cells and marked poikilocytosis including blister cells, spherocytes and echinocytes, consistent with oxidative hemolysis. High performance liquid chromatography (HPLC) showed a very conspicuous peak of methemoglobin (Fig. 2). Direct Coombs test and the paroxysmal nocturnal hemoglobinuria screen using DiaMed-ID card were negative. The overall picture was consistent with an active hemolysis due to G6PD deficiency.
A serial of ABGs showed gradual increment of the met-Hb level but methylene blue could not be administered due to his low G6PD levels. He was managed with 100% oxygen, generous hydration and the broad spectrum antibiotic meropenem. However, his Hb gradually drop to 8 g/dL within 12 hours of admission, with increased LDH levels to 1639 U/L. In addition, his creatinine increased to 219 µmol/L despite aggressive hydration (120 mL/hr following the initial 1 liter bolus). He was transfused one unit of packed red blood cells, following which he developed pulmonary edema necessitating reduction of fluids to 83 mL/hr while continuing urine alkalization. Despite these measures, he continued to have deteriorating renal function with increased creatinine upto 300 µmol/L and worsening of pulmonary edema. Intravenous fluids were stopped and he was given diuretics and was started on non-invasive ventilation (NIV) and hemodialysis. During dialysis, he developed asystole and passed away despite resuscitative measures.
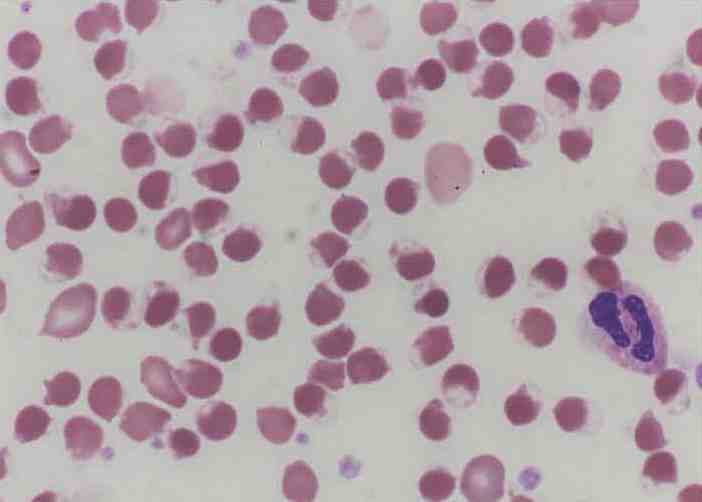
Figure 1: Blood film showing numerous blister cells indicating oxidative hemolysis.
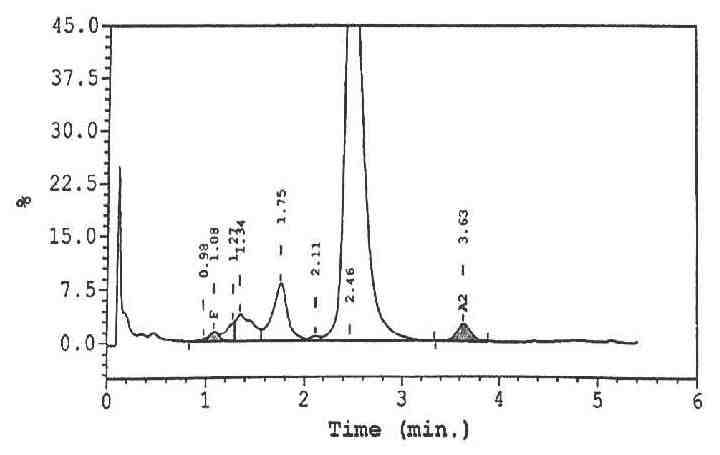
Figure 2: HPLC showing a fast peak at the left most region of the chromatograph representing the methemoglobin
Discussion
G6PD deficiency is common in Oman with the G6PD Mediterranean mutation accounting for most cases.2 The case presented herein has many unusual features. First, it is a late presentation of G6PD deficiency. No test for congenital causes of met-Hb was done since these causes are very unusual to present at such a late age, even with the class II variant. Second, the factor triggering such hemolysis remains unknown. Infection is probably the most common factor inciting hemolysis in G6PD-deficient patients. Although the patient had a very high WBC and fever, he was not toxic-looking and subsequent cultures were all negative. In addition, he was covered with broad spectrum antibiotics. Relevant medications and other precipitating factors were ruled out. However, potential toxins could not be ruled out due to the lack of necessary tests at the local hospital.
The presence of met-Hb should be suspected when the oxygen saturation as measured by pulse oximetry is significantly different from the oxygen saturation calculated from arterial blood gas analysis (saturation gap),8 as evident in this case report. Met-Hb is usually treated with 100% oxygen and 1% methylene blue solution. The methylene blue acts as an electron acceptor for NADPH met-Hb reductase. NADPH is then generated via the pentose phosphate pathway. Methylene blue however, is contraindicated in G6PD deficiency since the reduction of met-Hb by methylene blue is dependent upon NADPH generated by G6PD. As a result, methylene blue may not only be ineffective but is also potentially dangerous, since it has an oxidant potential that may induce hemolysis in G6PD deficient subjects.9 If methylene blue is contraindicated, moderate doses of ascorbic acid (300 to 1000 mg/day orally in divided doses) should be given, as it may also cause oxidant hemolysis in G6PD-deficient patients when given in very high doses.
Conclusion
Methemoglobinemia may present in patients with G6PD deficiency and severe hemolysis and treating it with methylene blue may be detrimental. Search and treatment for the root cause is mandatory. G6PD deficiency may present late in life especially in areas with mild variants.
Acknowledgements
The authors reported no conflict of interest and no funding was received for this work.
References
1. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008 Jan;371(9606):64-74.
2. Daar S, Vulliamy TJ, Kaeda J, Mason PJ, Luzzatto L. Molecular characterization of G6PD deficiency in Oman. Hum Hered 1996 May-Jun;46(3):172-176.
3. Raupp P, Hassan JA, Varughese M, Kristiansson B. Henna causes life threatening haemolysis in glucose-6-phosphate dehydrogenase deficiency. Arch Dis Child 2001 Nov;85(5):411-412.
4. Clark BB, Morrissey RW. Relation of methemoglobin to hemolysis. Blood 1951 Jun;6(6):532-543.
5. Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011 Nov;104(11):757-761.
6. Taber KH, Migliore PJ, Pagani JJ, Ford JJ, McLauren T, Hayman LA. Temporal changes in the oxidation state in in vitro blood. Invest Radiol 1990 Mar;25(3):240-244.
7. Hamirani YS, Franklin W, Grifka RG, Stainback RF. Methemoglobinemia in a young man. Tex Heart Inst J 2008;35(1):76-77.
8. Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004 Sep;83(5):265-273.
9. Rosen PJ, Johnson C, McGehee WG, Beutler E. Failure of methylene blue treatment in toxic methemoglobinemia. Association with glucose-6-phosphate dehydrogenase deficiency. Ann Intern Med 1971 Jul;75(1):83-86.
|