| |
Abstract
Among the inherited bone marrow failure disorders, dyskeratosis congenita is an X-linked inherited disorder arising as a consequence of short telomere and mutations in telomere biology. Production of the altered protein dyskerin, leads to vulnerable skin, nails, and teeth which lead to higher permeability for noxious agents which can induce carcinogenesis accounting for the classical triad of skin pigmentation, nail dystrophy and oral leukoplakia. This condition is fatal and patients succumb to aplastic anemia, malignancy or immunocompromised state. We present a young male with the classic clinical triad and avascular necrosis of both femoral heads, with no evidence of hematologic anomaly or any malignancy. He was managed for osteonecrosis with uncemented total hip arthroplasty for the symptomatic left hip. Our case represents a benign form of such a fatal and rare condition, which if detected and managed early can result in improved quality of life for the patient suffering from this disorder. This patient is under our meticulous follow-up for the last 2 years in order to determine any late development of complications before being labelled as a variant of this syndrome.
Keywords: Dyskeratosis congenita; Osteonecrosis; Leukoplakia; Dyskerin; Inherited bone marrow failure syndrome.
Introduction
Dyskeratosis congenita (DC) is a rare, hereditary disease, which was first described by Zinsser in 1906, also known as Zinsser-Cole-Engman syndrome.1-3 DC occurs mostly in males and clinically manifests between 5 to 12 years. The classic triad of skin pigmentation, nail dystrophy and oral leukoplakia occur within complete expression of this syndrome. It is a fatal condition in which the majority of patients develop aplastic anemia, and malignant transformation occurs in the keratotic white patches.1-3 DC is a clinically heterogeneous inherited bone marrow failure syndrome (IBMFS), that epitomizes the clinical consequences of very short telomeres and mutations in telomere biology.4,5 As more patients with DC and their family members have been studied, an extremely broad clinical phenotype is now appreciated. Apart from the classical triad, additional features include epiphora, blepharitis, premature gray hair, alopecia, developmental delay, short stature, cerebellar hypoplasia, microcephaly, esophageal stenosis, urethral stenosis, pulmonary fibrosis, liver disease, avascular necrosis of hips (AVN) or shoulders, epithelial cancers, myelodysplastic syndrome (MDS), and leukemia.4-6 Hoyeraal-Hreidarsson (HH) syndrome is a severe form of DC with bone marrow failure, immunodeficiency, microcephaly, cerebellar hypoplasia, intrauterine growth retardation, and developmental delay.7,8 Revesz Syndrome, characterized by bilateral exudative retinopathy, bone marrow hypoplasia, nail dystrophy, fine hair, cerebellar hypoplasia, and growth retardation, also appears to be in the DC disease spectrum.9,10
We present a case of a young male patient with the classical clinical triad of DC with bilateral avascular necrosis (AVN) of both femoral head severely debilitated and wheelchair-bound due to left hip AVN-induced arthritis managed by an integrated approach of medical, dermatologic and orthopedic specialists. The patient was duly informed and he consented to the publication of his case report for medical benefit.
Case Report
A 22-year-old man presented to the orthopedic clinic with debilitating pain in the left hip since the last 7 months resulting in progressive limping and ambulating with a wheelchair. He was a bright software programmer and was out of work in view of his left hip problem for the last 7 months. He had a traumatic right femoral shaft fracture for which fixation was done with intramedullary nail 6 years back.The fracture has united. He had no history of alcoholism, smoking, chewing tobacco, or chronic drug intake. He had a history of hepatitis 4 years back which was managed symptomatically. He was observed to be having asymptomatic brownish skin pigmentation all over the body predominantly over the chest, trunk, arms, and thighs since birth; changes in the oral mucosa since birth; and a history of change in nail morphology since 8 years of age.
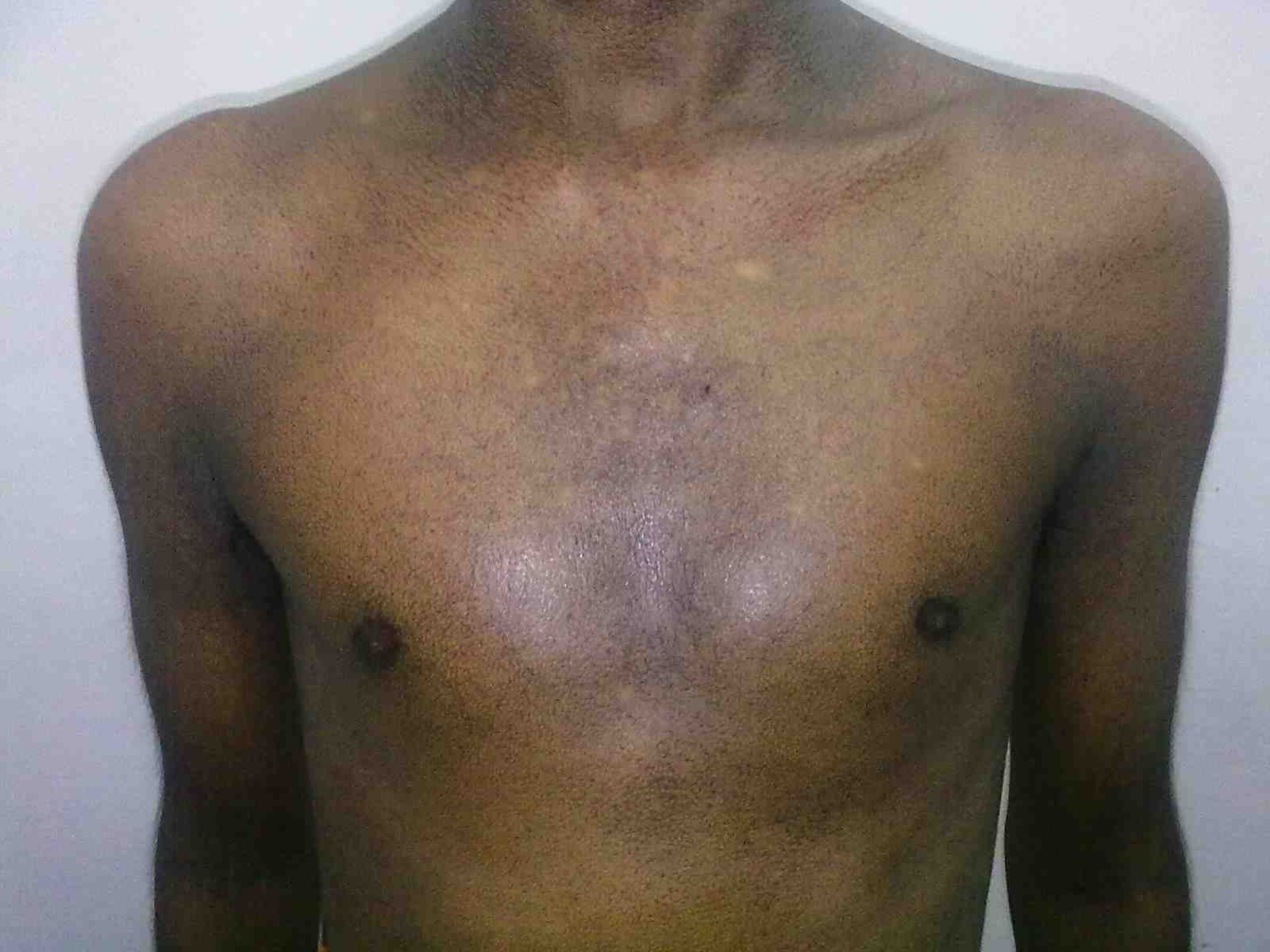
Figure 1: Fine reticulate greyish brown pigmentation over front of chest.

Figure 2: Dystrophy of nails of fin.
General examination showed fine, reticulate, grey brown pigmentation over the chest, abdomen, arms, thighs and back (Fig. 1). There was micrognathia and associated dystrophy in all the nails, (Figs. 2 and 3). Leukoplakia was present in the oral cavity forming irregular blackish patches on the buccal mucosa and tongue (Fig. 4). Examination of the hip revealed tenderness in both hips. There was fixed flexion deformity of 30 degrees in the left hip and painful restricted range of movements especially abduction and internal rotation on the left side. Limb length discrepancy of 5 cm was noted. Right hip was also mildly painful and having fixed flexion deformity of 15 degrees with painful range of movements. X-rays of pelvis and both hips revealed avascular necrosis of femoral head on both the sides, Ficat and Arlet stage IV on the left side with acetabular changes and stage II on the right side.11 (Fig. 5)
Hematology revealed hemoglobin of 14.2 gm% and normal blood counts. No clotting abnormality was noted. Liver function tests were normal except for a moderately elevated SGOT. The patient was planned for uncemented total hip replacement surgery on the left side. In view of his general skin and medical examination, a thorough medical, dermatologic and anesthetic evaluation were performed by respective specialists. Considering the symptoms of skin pigmentation, nail dystrophy and oral white patch, a differential diagnosis of pachyonychia congenita and dyskeratosis congenita were considered, and skin histopathological analysis was sent. After these exhaustive multidisplinary diagnostic evaluations, anesthetic clearance was obtained for the surgery. Uncemented left hip replacement surgery was performed under epidural anesthesia from a postero-lateral approach. The intraoperative and postoperative course was uneventful. Post-operatively, the patient was kept on below-knee skin traction, overcoming the fixed flexion deformity. There was residual limb length discrepancy. The patient’s postop hemoglobin was reduced to 7 gm% and thus was transfused 4 units of packed RBCs. There were no transfusion reactions and the patient was ambulated on a walker after 5 days postop. His stitches were removed after 2 weeks and he was aided by a physiotherapist for his independent gait training till 6 weeks after surgery. He was discharged from the hospital after 2 weeks.
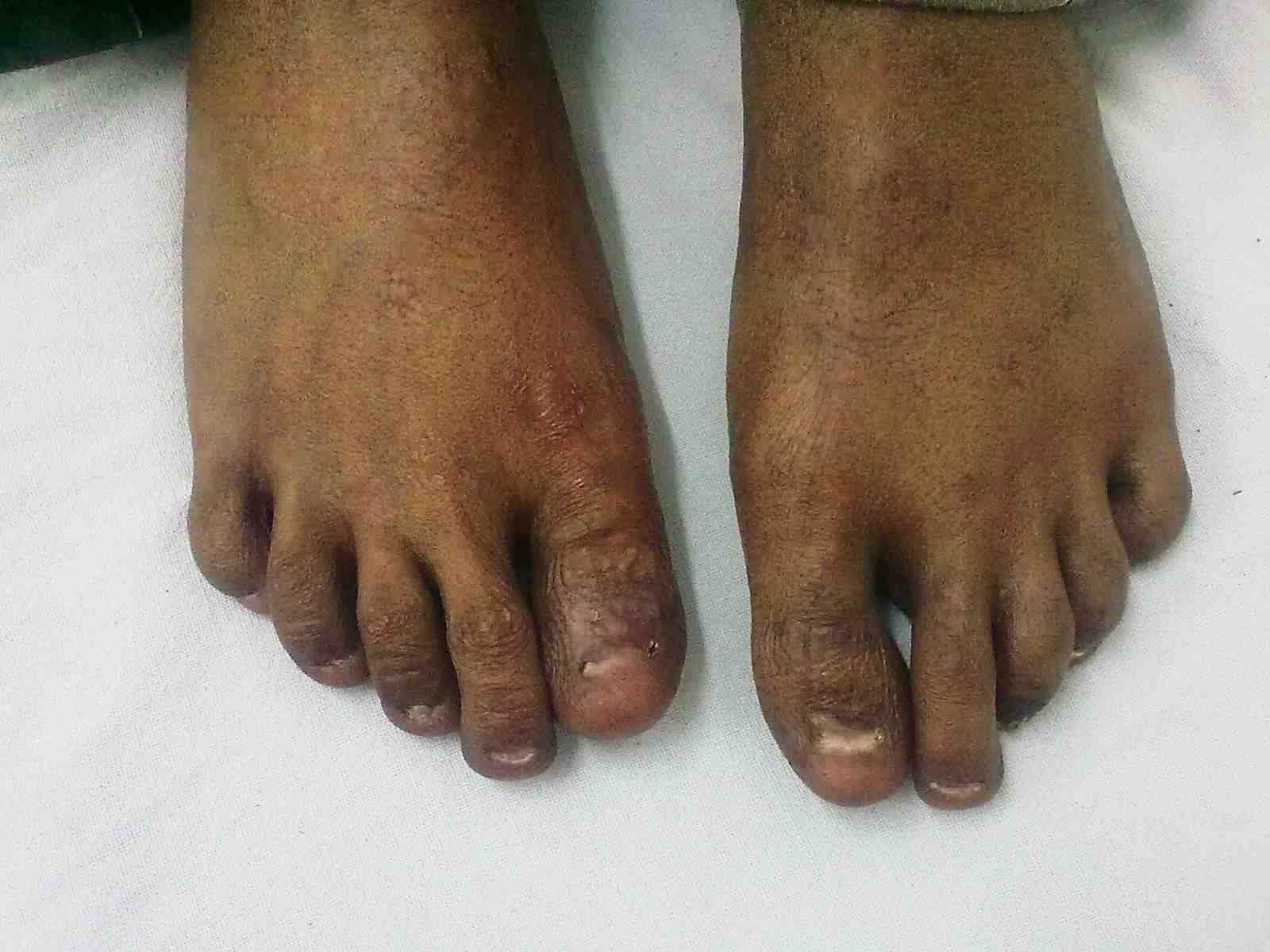
Figure 3: Dystrophy of toe nails.
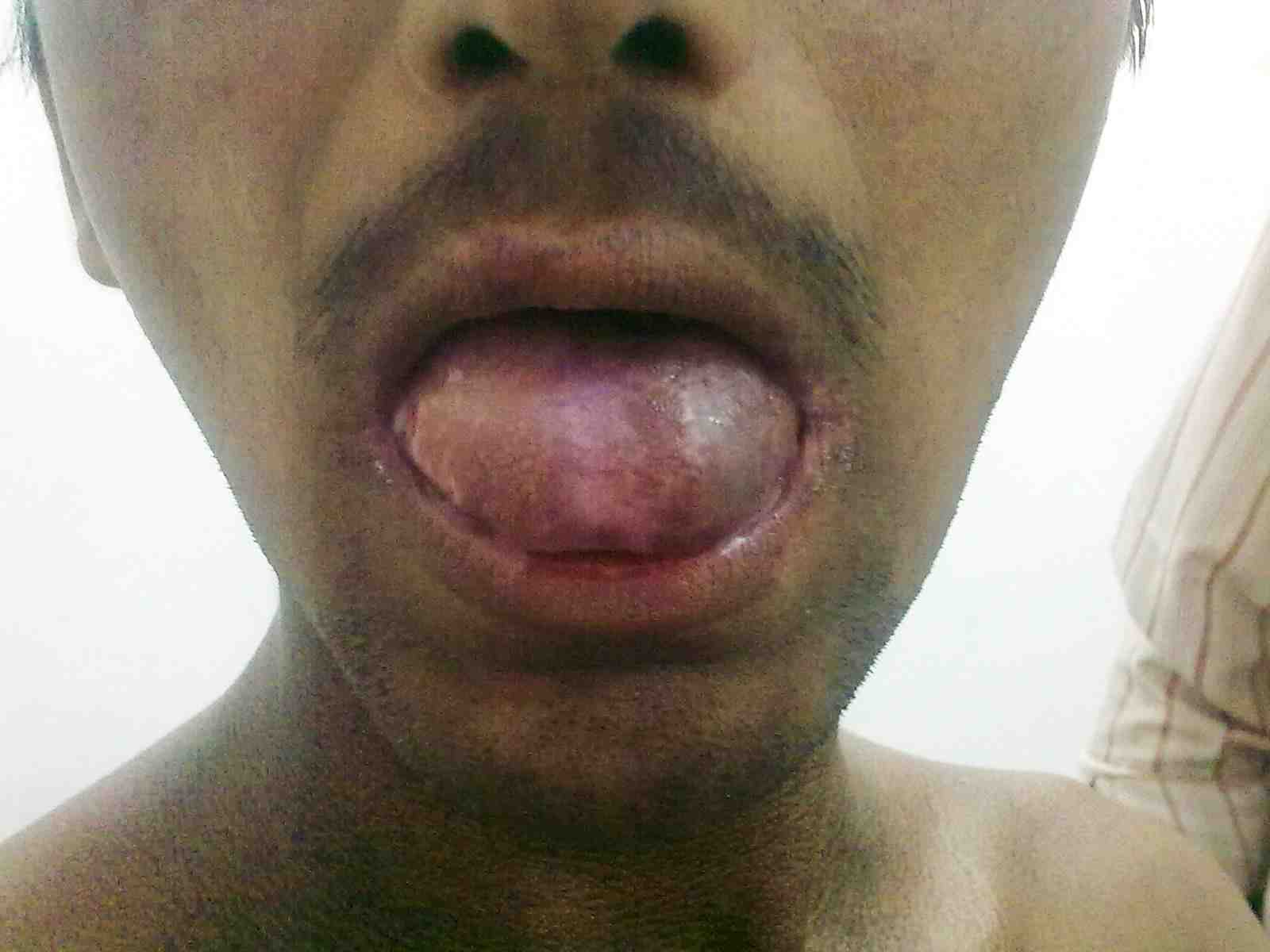
Figure 4: Oral leukoplakia.
Skin histopathology revealed markedly thinned out epidermis, exhibiting orthokeratosis and complete flattening of rete pegs. Basal layer showed focal vacuolation and increased amount of melanin pigment (Fig. 6). Dermis showed melanophages with extensive melanin incontinence and perivascular lymphocytic infiltrate. Histopathological report of the tongue lesion revealed hyperkeratosis with flattened rete ridges. There was no evidence of malignancy. The patient was prognosticated about the syndrome and its management and was advised about meticulous integrated follow-up (every 4 weeks till 2 years after surgery), with dermatologic and orthopedic specialists. At 2 years of his follow-up, he has done remarkably well orthopedically and is ambulating independently without any pain and is doing well in his previous occupation as a software programmer. His dermatologic symptoms are also constant and have not shown any worsening, he has been advised to follow up every 3 months.
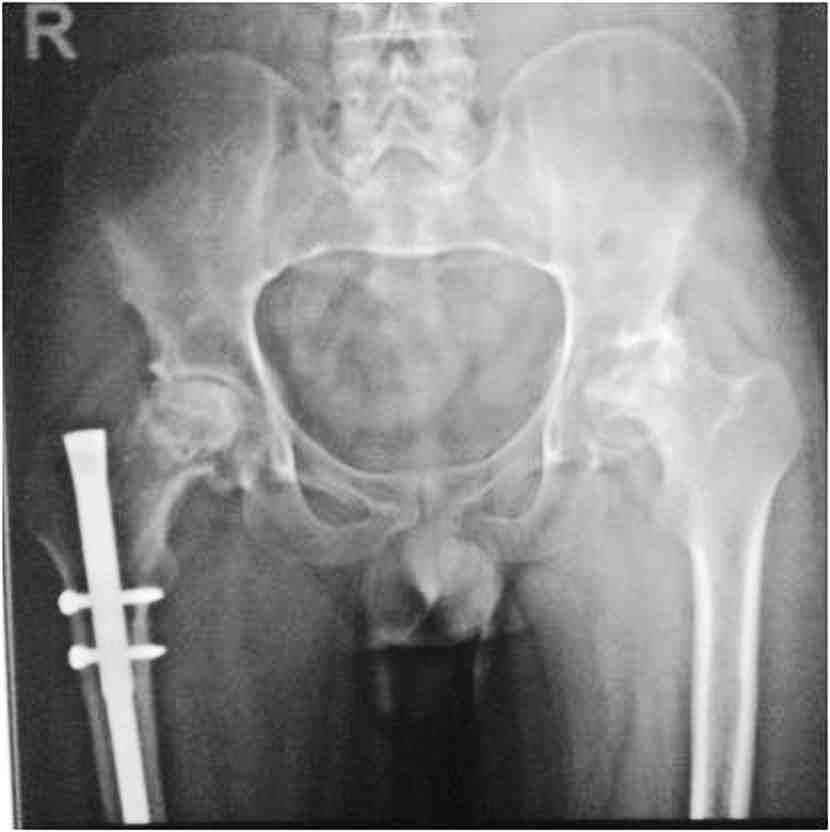
Figure 5: Preop X-ray pelvis with both hips showing osteonecrosis of both hips, left more advanced than right, with femur interlock nail in situ on right side.
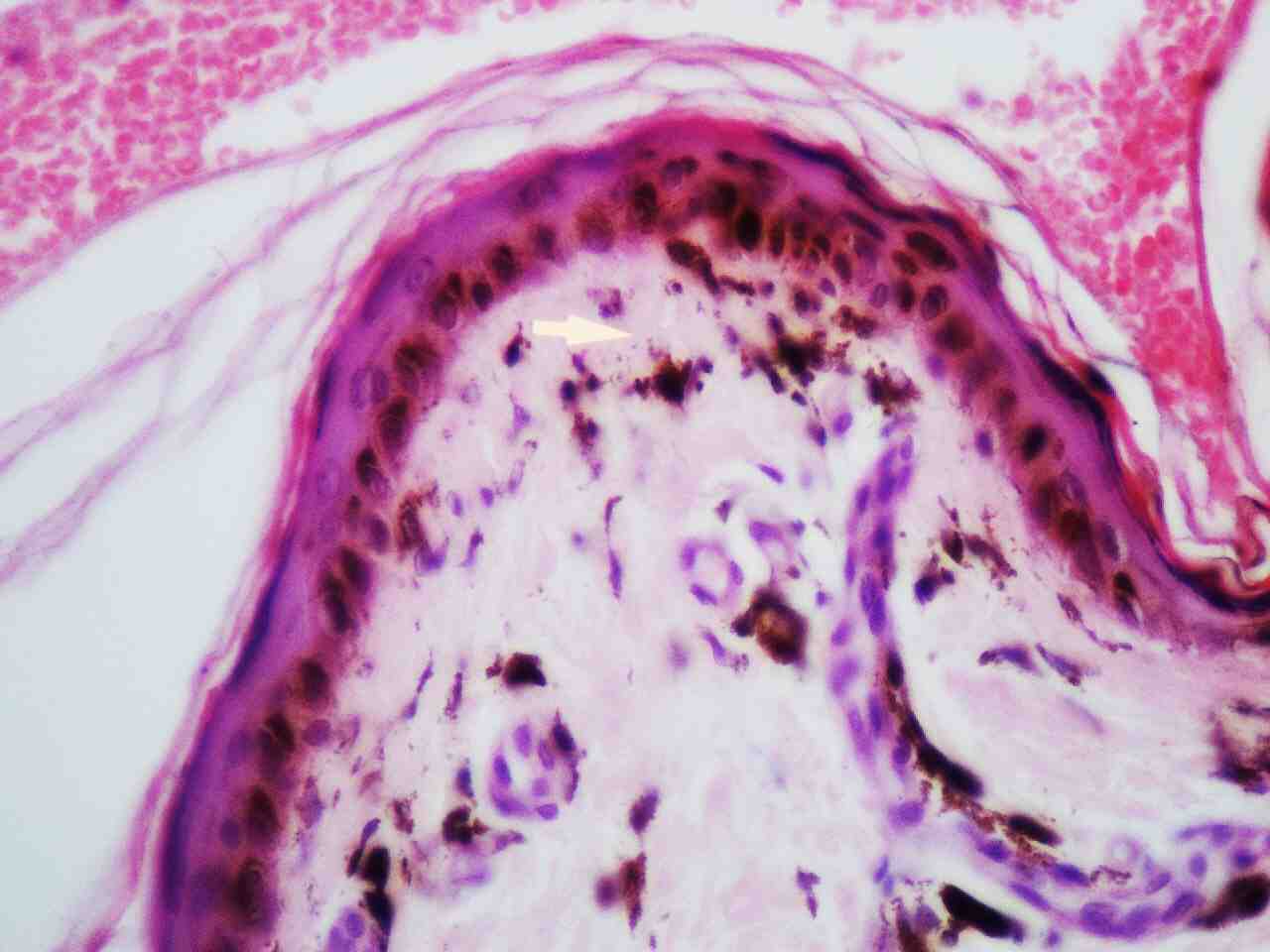
Figure 6: Skin histopathology shows epidermal atrophy, focal vacuolation of basal layer and melanin incontinence in the dermis (highlighted by an arrow).
Discussion
DC is reported to be a fatal condition with multisystem manifestations. There are X-linked, autosomal dominant and autosomal recessive forms of DC. It mainly occurs in males, inherited as X-linked recessive disorder with male:female predilection of 13:1.12 Occurrence in extremely low percentage of females suggests that there are subsets inherited in an autosomal dominant fashion.12 Autosomal dominant form of dyskeratosis congenita is associated with mutations in the RNA component of telomerase, hTERC, while X-linked dyskeratosis congenita is due to mutations in the gene encoding dyskerin, a protein implicated in both telomerase function and RNA processing.13 Intraorally, the white patch may affect the buccal mucosa, tongue or palate, with the tongue being most commonly affected as seen in our patient.
Electron microscopy studies revealed that cells in dyskeratosis congenita have an embryonic immature nucleus, which have higher chances to undergo malignant transformation.14 In addition, the barrier zone of epithelium is less effective in dyskeratosis congenita than the normal epithelium causing increased permeability of noxious substances and carcinogens to the germinal layers.15 Hence, increased malignant transformation rate is seen in leukoplakic areas associated with dyskeratosis congenita. However, in the case reported here, the biopsy report was not suggestive of any malignant changes, it just revealed markedly thinned out epidermis, exhibiting orthokeratosis, complete flattening of rete pegs. The basal layer showed focal vacuolation and increased amount of melanin pigment (Fig. 6). The dermis showed melanophages with extensive melanin incontinence and perivascular lymphocytic infiltrate.
In DC, severe periodontal destruction occur due to anomalies in ectodermally derived structures and diminished host response caused by neutropenia.15 Patients may have gingival inflammation, bleeding, recession, and bone loss that simulate juvenile periodontitis. In addition there may be defects in the structure of the enamel like hypocalcification that may cause dental caries. However, our patient had a healthy periodontium and his teeth were normal. Majority of patients develop a hematopoietic disorder resembling Fanconi’s anemia because initially the bone marrow is normal but gradually fat cells and fibrotic tissue replace the hematopoietic cells. Hematopoietic disorder manifests as marrow hypoplasia, anemia, or thrombocytopenia.16 With the development of pancytopenia, there occur secondary infections, causing death in the majority of these patients.
No opportunistic infections were seen in our patient. Other abnormalities like alopecia, taurodontism, mental retardation, small genitals, premature aging, nutmeg liver, horseshoe kidneys, amyloidosis, and development of Hodgkin’s disease and adenocarcinoma have also been reported,12,16 but not observed in this patient. The avascular necrosis (AVN) of bilateral femoral heads and humeral heads commonly associated with DC can lead to severe restriction of hip and shoulder joint function.4-6 Our patient presented with painful debilitating AVN-induced arthritis of the left hip which was managed with successful left hip replacement surgery restoring his normal function. The only curative therapy to date for a full blown case of DC has been bone marrow transplantation.3-5 However, the preparative regimens generally used during the bone marrow transplantation can adversely impact the susceptibility to malignancy. Steroids, granulocyte macrophage colony stimulating factors, and erythropoietin may be helpful transiently. Treatment with multiple cytokines (IL-3, IL-6) may offer additional benefits. Anti-thymocytic globulin (ATG) is not recommended for these patients. Gene therapy may become a feasible consideration.
Once an index case has been identified, genetic counselling is important. The parents must be oriented to the pattern of inheritance and the prospect of prenatal diagnosis. This will allow early harvest and storage of their bone marrow for use after anticipated marrow failure. In our patient, fortunately, there was no worsening of bone marrow functions and he has done remarkably well medically and orthopedically after 2 years of his diagnosis. We believe that he requires further meticulous follow-up to look for any late development of complications before being labelled as a variant of this syndrome.
Conclusion
Our case presents with probably the most benign presentation of this rare and serious disorder to be reported in the medical literature. The unique features of this case were the presentations of major orthopedic problem, skin manifestations with no anomaly of gingival, and hematopoiesis. We want to highlight the unusual manifestation of the syndrome and recommend close multidisciplinary follow-up of such cases for progression to malignancies of blood through regular blood count, and skin and bone marrow biopsy as necessary.
Acknowledgements
Authors reported no conflict of interest and no funding was received for this work.
References
1. Tanaka A, Kumagai S, Nakagawa K, Yamamoto E. Cole-Engman syndrome associated with leukoplakia of the tongue: a case report. J Oral Maxillofac Surg 1999 Sep;57(9):1138-1141.
2. De Boeck K, Degreef H, Verwilghen R, Corbeel L, Casteels-Van Daele M. Thrombocytopenia: first symptom in a patient with dyskeratosis congenita. Pediatrics 1981 Jun;67(6):898-903.
3. Shetty S, Gokul S. Keratinization and its disorders. Oman Med J 2012 Sep;27(5):348-357.
4. Walne AJ, Marrone A, Dokal I. Dyskeratosis congenita: a disorder of defective telomere maintenance? Int J Hematol 2005 Oct;82(3):184-189.
5. Yamaguchi H. Mutations of telomerase complex genes linked to bone marrow failures. J Nippon Med Sch 2007 Jun;74(3):202-209.
6. Drachtman RA, Alter BP. Dyskeratosis congenita: clinical and genetic heterogeneity. Report of a new case and review of the literature. Am J Pediatr Hematol Oncol 1992 Nov;14(4):297-304.
7. Sznajer Y, Baumann C, David A, Journel H, Lacombe D, Perel Y, et al. Further delineation of the congenital form of X-linked dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome). Eur J Pediatr 2003 Dec;162(12):863-867.
8. Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood 2006 Apr;107(7):2680-2685.
9. Kajtár P, Méhes K. Bilateral coats retinopathy associated with aplastic anaemia and mild dyskeratotic signs. Am J Med Genet 1994 Feb;49(4):374-377.
10. Revesz T, Fletcher S, al-Gazali LI, DeBuse P. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: a new syndrome? J Med Genet 1992 Sep;29(9):673-675.
11. Arlet J, Ficat RP. Forage-bioipsie de la tête fémorale dans l'ostéonécrose primative. In: Observations histo-pathologiques portant sur huit foranes. 2nd edn. Rev Rhumat. 31:1964;p. 257–264
12. Ogden GR, Connor E, Chisholm DM. Dyskeratosis congenita: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol 1988 May;65(5):586-591.
13. Bessler M, Wilson DB, Mason PJ. Dyskeratosis congenita and telomerase. Curr Opin Pediatr 2004 Feb;16(1):23-28.
14. McKay GS, Ogden GR, Chisholm DM. Lingual hyperkeratosis in dyskeratosis congenita: ultrastructural findings. J Oral Pathol Med 1991 Apr;20(4):196-199.
15. Yavuzyilmaz E, Yamalik N, Yetgin S, Kansu O. Oral-dental findings in dyskeratosis congenita. J Oral Pathol Med 1992 Jul;21(6):280-284.
16. Womer R, Clark JE, Wood P, Sabio H, Kelly TE. Dyskeratosis congenita: two examples of this multisystem disorder. Pediatrics 1983 Apr;71(4):603-609.
|
|