Infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD) is a rare autosomal recessive mitochondrial disorder first reported in 2014.1,2 IMNEPD is caused by various mutations in the peptidyl-tRNA hydrolase 2 (PTRH2) gene located on chromosome 17.1 This gene encodes a protein with hydrolase activity, which contributes to protein synthesis and plays a vital role in regulating cell function, including cell survival and anoikis, stress resistance, and muscle differentiation.3–5 Loss of PTRH2 function in human development leads to pro-survival signals loss, resulting in IMNEPD.4,5
IMNEPD is a multisystemic disorder with variable phenotypes. Its clinical manifestations include intellectual disability (ID), global developmental delay including motor and severe speech delay, ataxia, sensorineural hearing loss (SNHL), progressive cerebellar hypotrophy or atrophy, peripheral sensorimotor neuropathy, hypothyroidism, diabetes mellitus, exocrine pancreatic insufficiency (EPI), and liver dysfunction.4,6 IMNEPD diagnosis is confirmed by genetic testing and the management is supportive.6 Herein, we report a child with a novel mutation of PTRH2 born to a consanguineous Bahraini family.
Case report
A 12-year-old Bahraini male with an antenatal history of placental insufficiency, intrauterine growth retardation , and reduced fetal movements suspecting birth asphyxia was born via induced, vaginal delivery at term. His birth weight and head circumference were 2.3 kg and 31 cm, respectively (3rd percentile each). Examination revealed a right undescended testis. His parents are first-degree cousins [Figure 1].
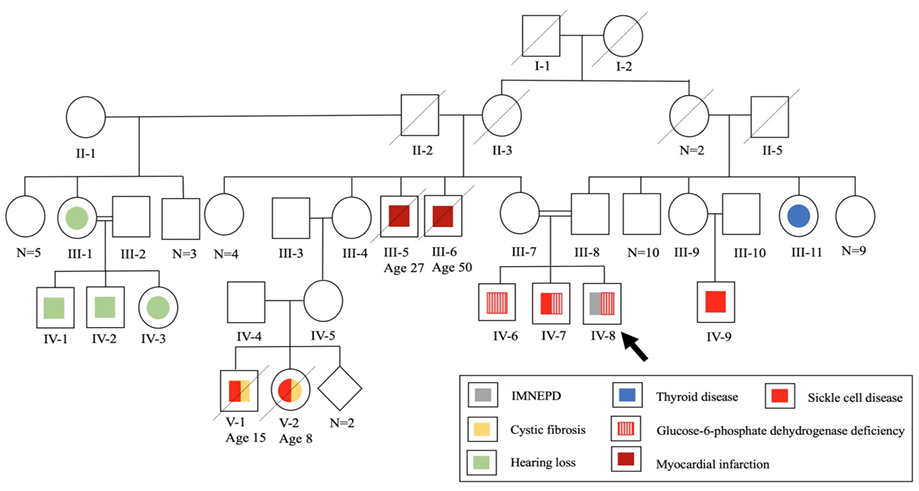 Figure 1: Family pedigree chart of a 12-year-old patient with infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD).
Figure 1: Family pedigree chart of a 12-year-old patient with infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD).
The patient developed constipation at two months. At four months, he was investigated for cystic fibrosis due to recurrent respiratory infections, abnormal bowel habits, and family history [Figure 1]. Sweat chloride test and fecal tryptic activity were normal and stool microscopy was negative for fat globules. Cow’s milk protein allergy was also suspected and managed.
At eight months, developmental delay and abnormal response to sounds were noted. Examination revealed hypotonia and a small head. His sibling's head circumference was also small (45 cm (< 1st percentile) at two years), raising the possibility of familial microcephaly. On re-evaluation at one year, the child could stand with support, sit, transfer objects, and babble, but could not crawl, or speak. Brain magnetic resonance imaging was normal. The child was assessed for speech and hearing difficulties, along with a positive family history [Figure 1]. Bilateral SNHL was confirmed, predominantly on the right side. Right cochlear implant surgery was performed, yet the patient remained nonverbal.
On follow-up, weight gain remained slow, and he developed steatorrhea with abdominal distension. Repeated sweat chloride test remained normal but stool was positive for fat globules. Celiac serology showed normal tissue transglutaminase but elevated antigliadin antibodies (AGA; immunoglobulin G (IgG) 70.7 U/mL (normal < 10 U/m)). Abdominal ultrasound (US) showed fatty liver while barium meal revealed gastroesophageal reflux disease with sliding hiatal hernia. Upper gastrointestinal (GI) endoscopy and histopathology confirmed gastroesophageal reflux disease.
At three years, a targeted familial mutation analysis (c.1911delG; p.Q637HfsX26 in exon 14 of cystic fibrosis transmembrane conductance regulator gene) was negative. Genitalia re-examination revealed normally descended testis.
The patient was lost to follow-up for years. At age 11, he was diagnosed with type 1 diabetes mellitus with diabetic ketoacidosis. He was referred to a gastroenterologist due to wasting and steatorrhea. On examination, he had subtle facial features with down slanting palpebral fissure, microcephaly, finger clubbing, and abdominal distention with gases [Figure 2]. He had an unstable, broad-based gait with a high steppage. Repeated celiac serology showed normal total IgA levels and AGA IgA, but high AGA IgG (26 U/mL) and tissue transglutaminase IgA (110 U/mL, normal: < 10 U/mL for all), and equivocal anti-endomysial antibody. Repeated upper GI endoscopy showed a longitudinal esophageal ulcer and unhealthy duodenal mucosa. Histopathology was compatible with acute esophagitis, chronic gastritis, and duodenitis.
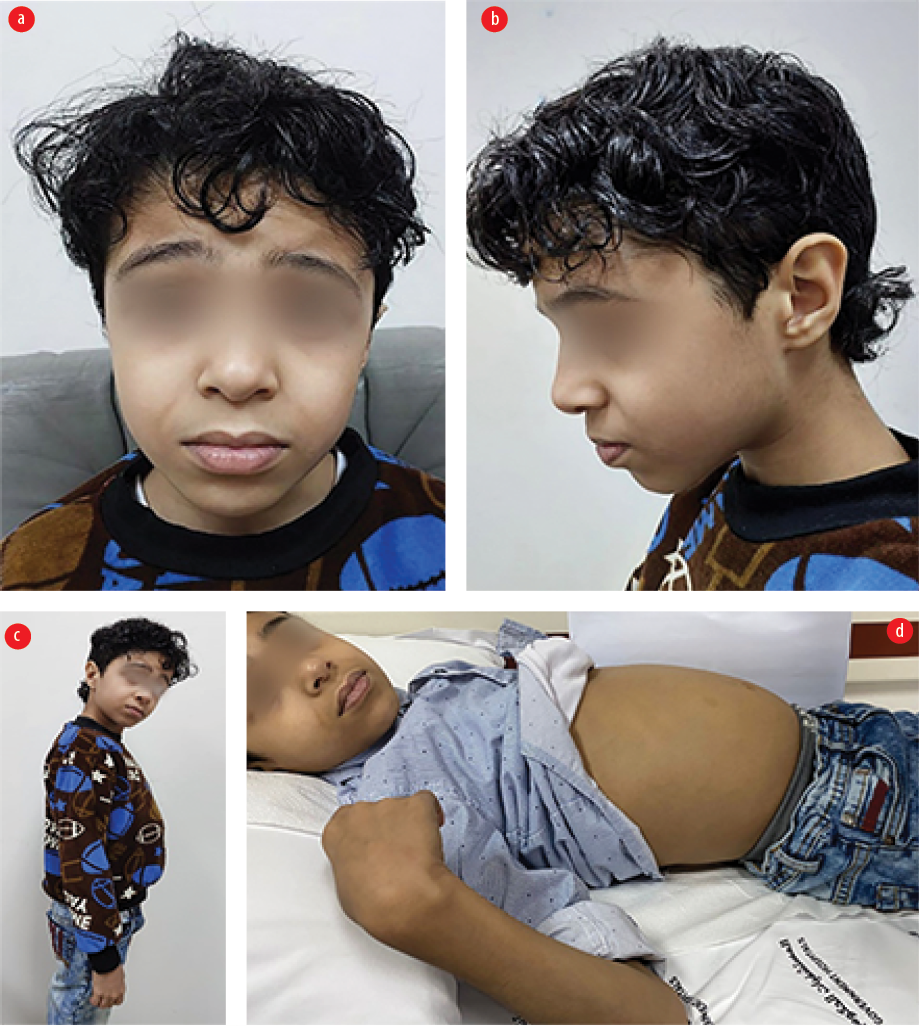 Figure 2: (a and b) Eleven-year-old patient showing subtle facial features with down slanting palpebral fissure and microcephaly along with (c and d) abdominal distension.
Figure 2: (a and b) Eleven-year-old patient showing subtle facial features with down slanting palpebral fissure and microcephaly along with (c and d) abdominal distension.
The whole exome sequencing (WES) test iden-tified a homozygous mutation (NM_001015509.2: c.370del p.(Glu124Lysfs*4)) in the PTRH2 gene. The PTRH2 variant c.370del p.(Glu124Lysfs*4) creates a shift in the reading frame starting at codon 124. This is consistent with autosomal recessive IMNEPD type 1. According to the American College of Medical Genetics and Genomics guidelines, the identified mutation is classified as a variant of uncertain significance (VUS). In addition, WES detected a heterozygous mutation in the hemoglobin subunit alpha 1 gene (NM_000558.3:c.95+2_95+6del) which is consistent with the patient’s known alpha-thalassemia carrier status. However, it did not report any genetic variants related to glucose-6-phosphate dehydrogenase (G6PD) or cystic fibrosis transmembrane conductance regulator genes, despite the patient’s known G6PD deficiency. This is because not all genetic variants detected by WES will be mentioned in the final report, especially if they are not relevant to the patient’s clinical history. Another possibility is that the detected G6PD mutation was a VUS, and in the absence of related clinical information, it was not reported. Both parents were heterozygous carriers for PTRH2 gene mutation.
At 12 years, an orthopedics review following foot trauma revealed waddling, foot drop, and distal muscle weakness. Muscular dystrophy was suspected. The patient is currently followed by a multi-disciplinary team providing supportive care. His last weight was 21.5 kg, height was 127 cm, and head circumference was 50 cm (all < 1st percentile) indicating severe thinness, short stature, and microcephaly, respectively.
Discussion
IMNEPD is a rare disease with a prevalence of < 1/1 000 000 (ORPHA:456312).7 A total of 19 patients with IMNEPD were reported worldwide. Our patient shared most of the clinical characteristics of the previously reported patients [Table 1].1,3,6,8–14 Of the 19 patients, 12 (63.1%) were males. The majority were children or adolescents (n = 16, 84.2%).
Table 1: Clinical features and genetic variants of 20 patients with IMNEPD published worldwide.
|
Patients, n
|
1
|
2
|
1†
|
5
|
3
|
3
|
1
|
1
|
1
|
2
|
1
|
|
Age, years
|
12
|
6, 14
|
10
|
3, 5, 7, 13, 15
|
13, 15, 17
|
7, 17, 27
|
12
|
30
|
19
|
16, 17
|
56
|
|
Sex
|
M
|
M, F
|
M
|
4M, 1F
|
3F
|
3M
|
F
|
M
|
F
|
M, F
|
M
|
|
Ethnicity
|
Bahraini
|
Turkish
|
Saudi
|
4 Saudi, 1 Tunisian
|
Arab
|
Syrian
|
Indian
|
Iranian
|
Indian
|
NR
|
Japanese
|
|
Consanguinity
|
+
|
+
|
+
|
+
|
+
|
+
|
-
|
NR
|
+
|
NR
|
-
|
|
homozygous PTRH2 variant
|
c.370del p.(Glu124Lysfs*4)
|
c.269_270delCT p.Ala90Glyfs*13
|
c.254A>C p.Q85P
|
c.254A>C p.Q85P
|
c.254A>C p.Q85P
|
c.324G>A p.W108*
|
c.127dupA, p.Ser43LysfsTer11
|
c.68 T > C, p.Val23Ala
|
c.328G>T; p.Glu110* (+ KIF1A c.223C > T, p.Arg75Trp, heterozygous)
|
c.127dupA, p.(Ser43Lysfs*11
|
c.280 T > A, p.Tyr94Asn,
|
|
Neurologic
|
|
|
|
|
|
|
|
|
|
|
|
|
Motor/speech delay
|
+
|
+
|
+
|
+
|
+
|
+
|
-
|
+
|
+
|
+
|
-
|
|
SNHL
|
+
|
+
|
+
|
+
|
+
|
+ (1/3)
|
+
|
-
|
+
|
+
|
+
|
|
Intellectual disability
|
+
|
+
|
+
|
+ (4/5) NR (1/5)
|
-
|
+
|
+
|
-
|
+
|
+
|
+
|
|
Post-natal microcephaly
|
-
|
+
|
NR
|
-
|
+
|
+
|
-
|
NR
|
-
|
+
|
NR
|
|
Hypotonia
|
+
|
+
|
+
|
+ (1/5)
|
+
|
+ (2/3)
|
-
|
NR
|
+
|
NR
|
NR
|
|
Distal muscle weakness
|
+
|
+
|
NR
|
+ (3/5)
|
+
|
+ (2/3)
|
-
|
-
|
+
|
NR
|
+
|
|
Ataxia
|
+
|
+
|
+
|
+ (4/5)
|
-
|
+
|
-
|
NR
|
+
|
+
|
+
|
|
Cerebellar hypoplasia/atrophy
|
-
|
+
|
NR
|
+ (1/5)
|
-
|
+ (2/3)
|
-
|
NR
|
+
|
NR
|
+
|
|
Peripheral neuropathy
|
+
|
+
|
NR
|
+ (1/5)
|
+
|
+ (1/3)
|
+
|
-
|
+
|
+
|
+
|
|
Facial palsy
|
-
|
+
|
NR
|
+ (2/5)
|
NR
|
NR
|
NR
|
-
|
-
|
NR
|
NR
|
|
Abnormal gait
|
+
|
-
|
NR
|
NR
|
+
|
+ (2/3)
|
NR
|
+
|
+
|
NR
|
+
|
|
Seizures
|
-
|
-
|
NR
|
NR
|
NR
|
+
|
-
|
+
|
+ one episode
|
NR
|
NR
|
|
EEG abnormality
|
Not done
|
+
|
NR
|
- (1/5)
NR (4/5)
|
NR
|
+ (2/3)
|
NR
|
NR
|
-
|
NR
|
NR
|
|
Face
|
|
|
|
|
|
|
|
|
|
|
|
|
Dysmorphic features
|
+
|
+
|
-
|
+ (3/5)
|
NR
|
+ (2/3)
|
-
|
-
|
-
|
NR
|
+
|
|
Myopia
|
-
|
NR
|
NR
|
NR
|
+
|
NR
|
NR
|
+
|
-
|
NR
|
NR
|
|
Endocrine
|
|
|
|
|
|
|
|
|
|
|
|
|
Diabetes mellitus
|
+
|
+
|
NR
|
+ (1/5)
|
NR
|
+ (2/3)
|
+
|
NR
|
+
|
+ (1/2)
|
-
|
|
Hypothyroidism
|
-
|
+
|
-
|
-
|
-
|
-
|
-
|
NR
|
-
|
+ (1/2)
|
+
|
|
Delayed menarche
|
NA
|
NR
|
NA
|
NR (1/5) NA (4/5)
|
+
|
NA
|
-
|
NA
|
+
|
NR
|
NA
|
|
Abdomen
|
|
|
|
|
|
|
|
|
|
|
|
|
Exocrine pancreatic insufficiency
|
+
|
+
|
NR
|
+ (3/5) NR (2/5)
|
-
|
+ (2/3)
|
-
|
-
|
+
|
+ (1/2)
|
-
|
|
Abnormal pancreas imaging
|
Not visualized
|
+ (1/2)
|
NR
|
+ (1/5)
|
NR
|
NR
|
-
|
NR
|
+
|
+ (1/2)
|
+
|
|
Hepatomegaly
|
-
|
+ (1/2)
|
NR
|
+ (1/5)
|
NR
|
-
|
-
|
-
|
NR
|
NR
|
-
|
|
Abnormal liver imaging
|
+
|
+
|
NR
|
+ (1/5)
|
-
|
NR
|
-
|
NR
|
+
|
NR
|
-
|
|
Genitourinary
|
|
|
|
|
|
|
|
|
|
|
|
|
Undescended testes or shawl scrotum
|
+ undescended testes
|
+ (1/2) Shawl scrotum
|
+ Undescen-ded testes
|
- Shawl scrotum (3/5) NR (2/5)
|
NA
|
+ (1/3) Undescended testes
|
NA
|
NR
|
NA
|
NR
|
NR
|
|
Skeletal
|
|
|
|
|
|
|
|
|
|
|
|
|
Hand deformity
|
-
|
+ (2/2)
|
NR
|
+ (3/5)
|
-
|
+ (1/3)
|
-
|
-
|
-
|
+
|
+
|
|
Foot deformity
|
-
|
+ (1/2)
|
+
|
+ (4/5)
|
+ (1/3)
|
+ (1/3)
|
-
|
+
|
+
|
NR
|
+
|
+: present; -: not present; NR: not reported; NA: not applicable; SNHL: sensorineural hearing loss; EEG: electroencephalogram. †This child is one of the five patients reported by Picker-Minh et al.6
The proband’s history revealed placental insufficiency, reduced fetal movement, and suspected birth asphyxia. Similarly, a case with birth hypoxia,9 and another case with a history of reduced fetal movement have been reported.1
Figure 3 shows the clinical features reported in IMNEPD patients, including our patient.1,3,6,8–14 Subtle facial dysmorphism was noticed in our patient but no signs of skeletal deformity except for short stature at age 13. Several features of facial dysmorphism were described, including midface hypoplasia, hypertelorism, exotropia, and thin upper lip vermilion.1 Hands and feet are commonly affected, with features including proximal placement of thumbs, long fingers, ulnar deviation of the second and third fingers, abnormality of the hallux talipes, equinovalgus, and Achilles tendon contracture.1,6 Other features include congenital hip dislocation, seronegative spondyloarthropathy, and scoliosis.1,10,12 Our case was negative for postnatal microcephaly, yet microcephaly became prominent on follow-up. Microcephaly was also reported by other studies.1,3,9,13 Hu et al,1 found skeletal muscle fibrosis on the US in one patient, while Ando et al,14 described another patient with nonspecific changes of left biceps muscle biopsy. These investigations were not performed in our patient.
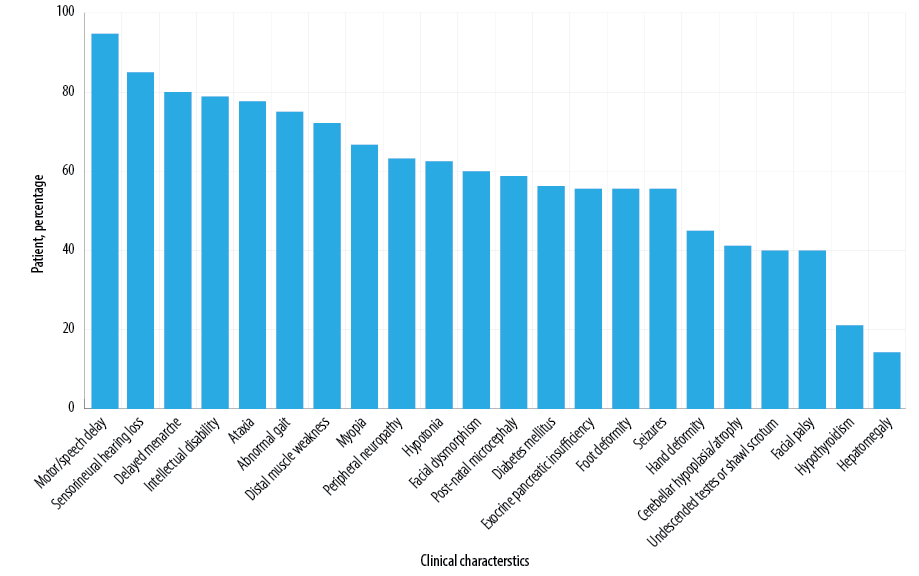 Figure 3: Clinical features in patients with infantile-onset multisystem neurologic, endocrine, and pancreatic disease.
Figure 3: Clinical features in patients with infantile-onset multisystem neurologic, endocrine, and pancreatic disease.
Neurological features in our patient included speech and motor delay, SNHL, and ID. ID was variable among reported cases.1,3,6 Ataxia, abnormal gait, distal muscle weakness, and hypotonia were frequently reported. Our patient had hypotonia and a broad-based gait with high steppage. After the foot trauma, he walked on inverted foot with waddling, foot drop, and muscle dystrophy. This indicated peripheral neuropathy. Similarly, Charles et al,12 reported one patient with a broad-based gait.Le et al,9 described two brothers, one with a broad-based gait with high steppage and the other with the same gait with mild truncal ataxia and hip girdle weakness. Sharkia et al,3 described three sisters with gait instability. Ando et al,14 reported a case with gait instability at 50 years, making him the only recorded adult-onset gait abnormality.
Progressive cerebellar hypoplasia/atrophy was identified in seven patients.1,6,9,12,14 Moreover, sensorimotor or sensorineural demyelinating polyneuropathy and facial palsy were identified in 10 and four patients, respectively.6,9,10,12,14 Unlike our case, five reported patients developed seizures, and some had electroencephalogram abnormalities.9,11,12
EPI was the most common GI manifestation reported [Figure 3], which was demonstrated in our case. Additionally, hyperechogenic pancreas was identified on the US in four patients with EPI. Fatty liver and hepatomegaly were detected in three and two of those patients, respectively.1,6,12 In our case, fatty liver was found at age two, yet repeated US showed normal liver echogenicity at age 11.
The proband was diagnosed with type 1 diabetes mellitus at age 11 which was an early diagnosis compared to other cases.1,6,9–12 Delayed menarche has been reported in the majority of female patients.3,6,10,12 Endocrine involvement can include hypothyroidism.1,14
Undescended testis was detected in two patients, while shawl scrotum was detected in four others.1,6,8,9 Urolithiasis was reported in one case.11
Multi-organ involvement necessitates a wide range of differential diagnoses.9 cystic fibrosis was the primary differential diagnosis in our case. Syndrome of short stature, microcephaly, and endocrine dysfunction is another.6 It comprises of microcephaly that develops prenatally, ataxia, endocrine dysfunction, and cerebral atrophy.6 Pearson syndrome, Cockayne syndrome, Johanson-Blizzard syndrome, and Mitochondrial acidosis, Encephalopathy, Lactate Acidosis, and Stroke (MELAS) may present with SNHL, ataxia, endocrine abnormalities, and liver and/or pancreas involvement.6 In adolescents with type 1 and late-onset diabetes mellitus, congenital rubella syndrome can be considered.10 Developmental delay, microcephaly, and SNHL can be also found in those patients.10 Furthermore, IMNEPD can overlap with progressive myoclonic epilepsies, spinocerebellar ataxias, or metabolic disorders.9 Table 2 elaborates distinguishing features of some syndromes.6
Table 2: Differential diagnoses of infantile-onset multisystem neurologic, endocrine, and pancreatic disease and their distinguishing features.
|
Johanson-Blizzard syndrome
|
Facial dysmorphism, urogenital defects
|
|
SSMED, Pearson syndrome, Cockayne syndrome, MELAS
|
Photosensitivity/dry skin
|
|
Pearson syndrome, Johanson-Blizzard syndrome
|
Blood count or bone marrow abnormalities
|
|
Pearson syndrome, MELAS
|
Metabolic acidosis
|
|
MELAS
|
Encephalopathy
|
SSMED: syndrome of short stature, microcephaly, and endocrine dysfunction; MELAS: mitochondrial acidosis, encephalopathy, lactate acidosis, and stroke.
The diagnosis of our patient was confirmed by genetic testing via the presence of a frameshift PTRH2 mutation. PTRH2 encodes a 179-residue mitochondrial protein which comprises a mitochondrial domain (amino acids 1–61) and a catalytic hydrolase domain (amino acids 63–179).4 Nonsense and frameshift mutations in PTRH2 produce a more severe phenotype compared to missense mutations where PTRH2 function is more preserved.3,6,9 Additionally, the same mutation of PTRH2 can have variable phenotype, suggesting variable expressivity.9 Our patient demonstrates a previously unreported novel nonsense mutation of PTRH2 (NM_001015509.2: c.370del p.(Glu124Lysfs*4)). This is the first IMNEPD case reported from Bahrain. Considering the high rate of consanguineous marriage in Bahrain and the surrounding region, prenatal diagnosis to detect genetic variants of such diseases is important.
Conclusion
IMNEPD has a wide phenotypic spectrum owing to the multi-systemic involvement, which makes the diagnosis challenging. Further research to understand the disease pathogenesis and presentations, and to explore treatment options is needed.
Disclosure
The authors declared no conflicts of interest. Informed consent was obtained from the patient’s mother.
references
- 1. Hu H, Matter ML, Issa-Jahns L, Jijiwa M, Kraemer N, Musante L, et al. Mutations in PTRH2 cause novel infantile-onset multisystem disease with intellectual disability, microcephaly, progressive ataxia, and muscle weakness. Ann Clin Transl Neurol 2014 Dec;1(12):1024-1035.
- 2. Kniffin CL, Vernon HJ. Neurologic, endocrine, and pancreatic disease, multisystem, infantile-onset 1; IMNEPD1. 2022 [cited 2022 September 30]. Available from: https://omim.org/entry/616263.
- 3. Sharkia R, Shalev SA, Zalan A, Marom-David M, Watemberg N, Urquhart JE, et al. Homozygous mutation in PTRH2 gene causes progressive sensorineural deafness and peripheral neuropathy. Am J Med Genet A 2017 Apr;173(4):1051-1055.
- 4. Corpuz AD, Ramos JW, Matter ML. PTRH2: an adhesion regulated molecular switch at the nexus of life, death, and differentiation. Cell Death Discov 2020 Nov;6(1):124.
- 5. Doe J, Kaindl AM, Jijiwa M, de la Vega M, Hu H, Griffiths GS, et al. PTRH2 gene mutation causes progressive congenital skeletal muscle pathology. Hum Mol Genet 2017 Apr;26(8):1458-1464.
- 6. Picker-Minh S, Mignot C, Doummar D, Hashem M, Faqeih E, Josset P, et al. Phenotype variability of infantile-onset multisystem neurologic, endocrine, and pancreatic disease IMNEPD. Orphanet J Rare Dis 2016 Apr;11(1):52.
- 7. Orphanet: infantile multisystem neurologic endocrine pancreatic disease. 2022 [cited 2022 September 29]. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=456312.
- 8. Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep 2015 Jan;10(2):148-161.
- 9. Le C, Prasad AN, Rupar CA, Debicki D, Andrade A, Prasad C. Infantile-onset multisystem neurologic, endocrine, and pancreatic disease: case and review. Can J Neurol Sci 2019 Jul;46(4):459-463.
- 10. Parida P, Dubbudu A, Biswal SR, Sharawat IK, Panda PK. Diabetes mellitus in an adolescent girl with intellectual disability caused by novel single base pair duplication in the PTRH2 gene: expanding the clinical spectrum of IMNEPD. Brain Dev 2021 Feb;43(2):314-319.
- 11. Khamirani HJ, Zoghi S, Dianatpour M, Jankhah A, Tabei SS, Mohammadi S, et al. A novel PTRH2 missense mutation causing IMNEPD: a case report. Hum Genome Var 2021 Jun;8(1):23.
- 12. Charles Bronson S, Suresh E, Stephen Abraham Suresh Kumar S, Mythili C, Shanmugam A. A novel synergistic association of variants in PTRH2 and KIF1A relates to a syndrome of hereditary axonopathy, outer hair cell dysfunction, intellectual disability, pancreatic lipomatosis, diabetes, cerebellar atrophy, and vertebral artery hypoplasia. Cureus 2021 Feb;13(2):e13174.
- 13. Becker M, Seneca S, Schierloh U, Witsch M, De Beaufort C, Scalais E. Diabetes in a child with infantile onset multisystem neurological, endocrine and pancreatic disease (IMNEPD). Horm Res Paediatr 2021;94(Suppl 1):228-228.
- 14. Ando M, Higuchi Y, Takeuchi M, Hashiguchi A, Takashima H. The first case of infantile-onset multisystem neurologic, endocrine, and pancreatic disease caused by novel PTRH2 mutation in Japan. Neurol Sci 2022 Mar;43(3):2133-2136.