| |
Abstract
Infantile Systemic Hyalinosis (ISH) (OMIM 236490) is a rare, progressive and fatal autosomal recessive disorder characterized by multiple subcutaneous skin nodules, gingival hypertrophy, osteopenia, joint contractures, failure to thrive, diarrhea with protein losing enteropathy, and frequent infections. There is diffuse deposition of hyaline material in the skin, gastrointestinal tract, muscle and endocrine glands. It is caused by mutations in the ANTXR2 (also known as CMG2) gene, which encodes a trans-membranous protein involved in endothelial development and basement membrane-extracellular matrix assembly. We describe a child with classical features of ISH presenting in infancy with severe chronic debilitating pain and progressive joint contractures. The diagnosis was confirmed by molecular DNA sequencing of ANTXR2 gene which revealed a novel homozygous mutation not previously reported; 79 bp deletion of the entire exon 11 (c.867_945del, p.E289DfsX22). Although this is the first reported case of ISH in Oman, we believe that the disease is under-diagnosed since children affected with this lethal disease pass away early in infancy prior to establishing a final diagnosis.
Keywords: Infantile systemic hyalinosis; Joint contractures; Skin thickness.
Introduction
Infantile Systemic Hyalinosis (ISH) is a rare autosomal recessive condition characterized by progressive deposition of amorphous hyaline material in various tissues like skin, gastrointestinal tract, cardiac muscle, adrenals, skeletal muscles, lymph nodes, spleen, thyroid, and adrenal glands.1,2 It typically presents at birth or within the first few months of life with reduced spontaneous movements secondary to severe pain and progressive joint contractures. Progressive skin thickening, gingival hypertrophy, appearance of hyperpigmented patches over bony prominences and joints are the main dermatological manifestations.3 Hypotonia, osteopenia, bone fractures, short stature, persistent diarrhea with protein losing enteropathy (PLE) due to intestinal lymphangiectasia (IL), increased susceptibility to infections and failure to thrive are the main systemic manifestations.2,4-7 Children with ISH are cognitively normal. Susceptibility to infections and joints pain may become less severe if they survive infancy.
However, their mobility remains severely restricted by joint contractures.2 ANXR2 gene is the only gene known to be associated with ISH. In 2003, Dowling et al. and Hanks et al.2,8 simultaneously reported loss-of-function mutations in CMG2, on chromosome 4q21 in ISH. CMG2 may be responsible for basement-membrane matrix assembly and endothelial cell morphogenesis, suggesting that the hyaline material results from leakage of plasma components to the perivascular space.9 Majority of infants pass away before the age of two years as a result of multisystem failure.2 No specific treatment is currently available for this disorder, and is limited to symptomatic and supportive management as well as physiotherapy to maintain ambulation.4,9,10 Aggressive management was ineffective in one series.3 Use of d-penicillamine resulted in some improvement in joint mobility secondary to its inhibitory effect on collagen maturation was documented in a few reports.3 Subcutaneous nodules can be surgically excised, but the recurrence rate is high. Remarkable improvement using interferon-alpha 2b was reported in one series but this awaits confirmation.11
Case Report
A female baby was born at full term with birth weight of 3.3 kg to first degree consanguineous parents. There was a family history of three siblings (2 males, one female) dying in infancy with intractable diarrhea without clear diagnosis at different hospitals. This infant was noticed on the first day of life to have reduced spontaneous movements associated with severe pain with minimal handling. Within a few months, she developed progressive diarrhea. At the age of 5 months, she presented with failure to thrive with severely restricted and painful passive and active movement of the limbs. Development, other than gross-motor, was age-appropriate.
On examination, she was small for her age, irritable and an apprehensive infant. She had tense shining skin mainly over bony prominences with skin thickening and hyperpigmentation that was progressively more evident with age, with appearance of well-demarcated dark-red nodules. Joint contractures affected proximal interphalangeal joints (Fig. 1), as well as bilateral wrist, knees and ankle contractures that were noticed to be progressive with time. Other features included perioral papular skin rash (Fig. 2), gum hypertrophy and erythematous perianal skin tags. A clinical impression of ISH was suspected. Laboratory work-up was consistent with rickets (high alkaline phosphatase and parathyroid hormone and low phosphate levels), in addition to hypoalbuminemia and high fecal alpha 1 anti trypsin indicating protein losing enteropathy (PLE). There was radiological evidence of generalized osteopenia with bilateral distal femur green-stick fractures on skeletal X rays. (Figs. 3 and 4)
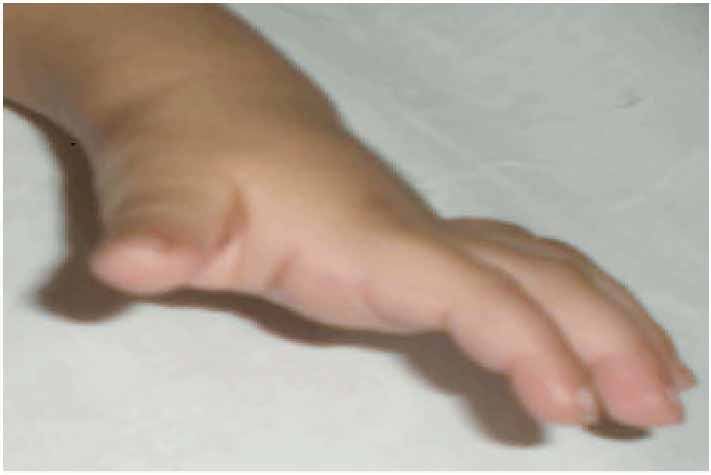
Figure 1: Contractures involving interphalengeal joints with mild thickening of the overlying skin. Skin over her digits was noted to be tight and shiny.
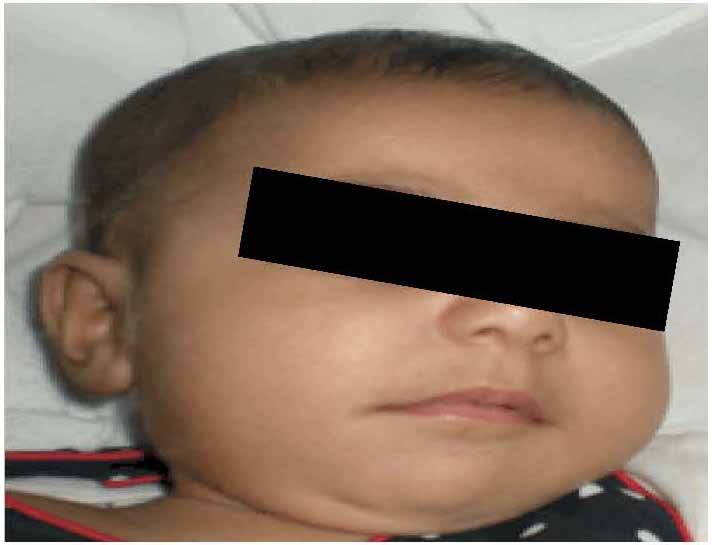
Figure 2: Patient at 5 months of age with mild perioral papular skin rash.
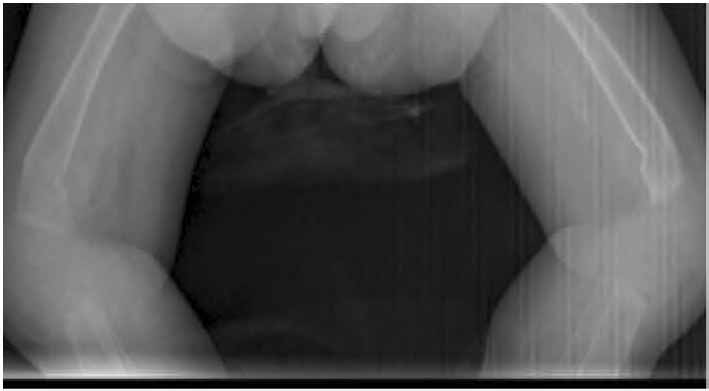
Figure 3: Bilateral green-stick fractures at the distal ends of both femoral bones.
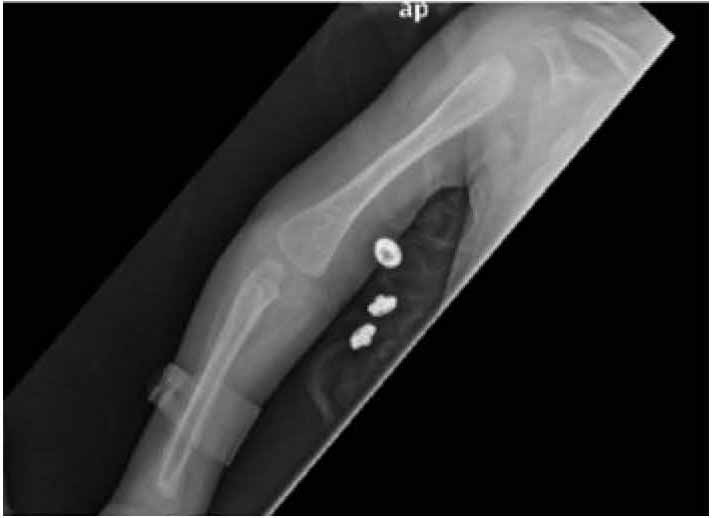
Figure 4: Marked osteopenia, thinning of the cortex and metaphyseal widening.
The child was managed conservatively with supportive care given with pain relief, physiotherapy, antimicrobial therapy, dietary modifications, albumin infusions and a trial of octreotide to reduce protein loss through possible intestinal lymphangiectasia described in this disorder. The child passed away at the age of 15 months with severe diarrhea and chest infection. Sequencing of ANTXR2 gene revealed a homozygous 79 bp deletion of the entire exon 11 (c.867_945del, p.E289DfsX22), confirming the diagnosis of ISH with a novel mutation. The parents were extensively counseled regarding the diagnosis and its grave outcome.
Discussion
This case report represents an infant who presented with typical clinical and biochemical features of ISH with a progressive and disabling joint pain and contractures leading to eventual death at age the of 15 months from severe chest infection and diarrhea. ISH was recognized as an entity after the first description by Landing and Nadorra in 1986.12 ISH is an autosomal recessive disorder that results from mutations in the anthrax toxin receptor 2 gene, ANTXR2.2,8,13 The ANTXR2 gene encodes capillary morphogenesis protein (CMG)2, a type 1 transmembrane protein. In skin and connective tissues, CMG2 binds to type IV collagen and laminins, and is considered a factor to basement membrane strength.12 Abnormal collagen metabolism and deposition causes skin abnormalities in ISH. Deficient extracellular matrix (ECM) synthesis and turnover is directly implicated in the pathogenesis of ISH.13 Skin biopsy in this patient was deemed unnecessary for diagnostic purposes since ANTXR2 gene mutation was identified.
To date, approximately 25 different pathogenic ANTXR2 mutations have been reported, comprising missense, nonsense and splice-site mutations.14,15 Majority of patients with ANTRX2 mutations were homozygous for private mutations with consanguinity being reported in a significant proportion of patients.3 The novel c.867_945del homozygous deletion in our patient resulted in a frame shift and a premature termination codon 22 amino acids downstream, p.E289DfsX22. This novel mutation is to be added to the described existing mutations in the literature. With our patient, having a novel mutation, there were no identified clinical or prognostic differences when compared to the reported cases worldwide. However, this first time described and valuable clinical and genetic finding could be of help in Omani (and perhaps in Arabs) patients suspected to have ISH as a screening and diagnostic tool. Management of this patient was carried out based on described case reports of supportive therapy. We started octreotide injections to minimize PLE in the patient without a significant difference seen in serum albumin levels. No conclusive effect could be drawn from the use octreotide in this patient because of the short period of trial prior to the patient's demise. We suggest that further studies be conducted to investigate the effect of octreotide in patients with ISH to diminish PLE.
Conclusion
The patient described in the current case report is the first diagnosed case in Oman with ISH to the best of our knowledge. The authors suspect the prevalence of ISH in Oman to be higher than reported; however, the disease might be under-recognized. This is supported by the fact that this child had 3 affected siblings who passed away in infancy without being diagnosed. The same conclusion was addressed in the largest case series of ISH.3 The group suggested that this is a common disorder in the Arab population in which they quoted a higher carrier frequency. To conclude, ISH is still a poorly understood entity of ground substance biology and poses a diagnostic as well as therapeutic dilemma for the treating physician. However, the major benefit in identifying pathogenetic mutations in individual cases of ISH is to make DNA-based prenatal diagnosis in subsequent pregnancies for couples with affected children.
Acknowledgements
The authors reported no conflict of interest and no funding was received on this work.
References
1. Shin HT, Paller A, Hoganson G, Willner JP, Chang MW, Orlow SJ. Infantile systemic hyalinosis. J Am Acad Dermatol 2004 Feb;50(2)(Suppl):S61-S64.
2. Hanks S, Adams S, Douglas J, Arbour L, Atherton DJ, Balci S, et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 2003 Oct;73(4):791-800.
3. Al-Mayouf SM, AlMehaidib A, Bahabri S, Shabib S, Sakati N, Teebi AS. Infantile systemic hyalinosis: a fatal disorder commonly diagnosed among Arabs. Clin Exp Rheumatol 2005 Sep-Oct;23(5):717-720.
4. Aghighi Y, Bahremand S, Nematollahi LR. Infantile systemic hyalinosis: report of three Iranian children and review of the literature. Clin Rheumatol 2007 Jan;26(1):128-130.
5. Glover MT, Lake BD, Atherton DJ. Infantile systemic hyalinosis: newly recognized disorder of collagen? Pediatrics 1991 Feb;87(2):228-234.
6. Parfitt AM. Familial neonatal hypoproteinaemia with exudative enteropathy and intestinal lymphangiectasis. Arch Dis Child 1966 Feb;41(215):54-62.
7. Büyükgebiz B, Oztürk Y, Arslan N, Ozer E. A rare cause of protein-losing enteropathy and growth retardation in infancy: infantile systemic hyalinosis. Turk J Pediatr 2003 Jul-Sep;45(3):258-260.
8. Al-Mubarak L, Al-Makadma A, Al-Khenaizan S. Infantile systemic hyalinosis presenting as intractable infantile diarrhea. Eur J Pediatr 2009 Mar;168(3):363-365.
9. Nézelof C, Letourneux-Toromanoff B, Griscelli C, Girot R, Saudubray JM, Mozziconacci P. [Painful disseminated fibromatosis (systemic hyalinosis): a new hereditary collagen dysplasia]. Arch Fr Pediatr 1978 Dec;35(10):1063-1074.
10. Ruiz-Maldonado R, Durán-McKinster C, Sáez-de-Ocariz M, Calderón-Elvir C, Yamazaki-Nakashimada MA, Orozco-Covarrubias L. Interferon alpha-2B in juvenile hyaline fibromatosis. Clin Exp Dermatol 2006 May;31(3):478-479.
11. Landing BH, Nadorra R. Infantile systemic hyalinosis: report of four cases of a disease, fatal in infancy, apparently different from juvenile systemic hyalinosis. Pediatr Pathol 1986;6(1):55-79.
12. Fong K, Rama Devi AR, Lai-Cheong JE, Chirla D, Panda SK, Liu L, et al. Infantile systemic hyalinosis associated with a putative splice-site mutation in the ANTXR2 gene. Clin Exp Dermatol 2012 Aug;37(6):635-638. Epub ahead of print.
13. Dowling O, Difeo A, Ramirez MC, Tukel T, Narla G, Bonafe L, et al. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 2003 Oct;73(4):957-966.
14. El-Kamah GY, Fong K, El-Ruby M, Afifi HH, Clements SE, Lai-Cheong JE, et al. Spectrum of mutations in the ANTXR2 (CMG2) gene in infantile systemic hyalinosis and juvenile hyaline fibromatosis. Br J Dermatol 2010 Jul;163(1):213-215.
15. Deuquet J, Abrami L, Difeo A, Ramirez MC, Martignetti JA, van der Goot FG. Systemic hyalinosis mutations in the CMG2 ectodomain leading to loss of function through retention in the endoplasmic reticulum. Hum Mutat 2009 Apr;30(4):583-589.
|
|