Trisomy 13 (T13) or Patau’s syndrome is an extremely rare but serious aneuploidy that occurs in 1 in 12 000 births, with an 86–91% mortality rate within the first year after birth. It results from either nondisjunction or Robertsonian translocation.1 The most common malformations are median facial or central nervous system defects, and the majority of those with T13 also suffer from congenital heart defects.1 About 25% of T13 patients are also affected by another genetic condition, Meckel’s diverticulum (MD), which almost always remains asymptomatic. Clinical presentation of MD in T13 neonates is extremely rare.2,3 Only two symptomatic cases of infants with T13 and MD are described in the literature. Here, we report a case of a full-term neonate with T13 and MD who presented with bilious aspirates and intermittent volvulus due to MD with band.
Case report
A full-term male infant (3671 g, 41 + 2 weeks gestation), was delivered to a 27-year-old prima-gravida by normal vaginal delivery. The neonate had Apgar scores of 6 and 8 at 1 and 5 minutes, respectively. The pregnancy had been normal, with normal prenatal serological results and normal antenatal ultrasound results in the second trimester. The parents were second-degree consanguineous. On physical examination, the baby was noticed to have multiple congenital anomalies including complete bilateral cleft lip/palate, low-set ears, microphallus, and bilateral undescended testes. Further evaluation after admission to neonatal intensive care unit revealed more anomalies. Cranial ultrasound revealed vermian hypoplasia and cystic malformation of the posterior fossa. An echocardiography showed moderate patent ductus arteriosus (PDA), small atrial septal defect (ASD), and bicuspid aortic valve. The newborn was intubated and mechanically ventilated to mitigate respiratory distress. The abdomen felt soft on palpation and appeared not-distended, with normal passage of meconium. Feeds were initiated at 5 mL every three hours on day two of life. Feeds were tolerated well for two days but on the third day, greenish aspirates were noted. The abdomen remained soft and non-distended. An upper gastrointestinal (GI) contrast study showed the duodenojejunal flexure to be in a normal position, but the upper bowel loops had a corkscrew appearance raising concern for volvulus [Figures 1 and 2].
.png)
.png) Figure 1: Fluoroscopic image of the upper gastrointestinal contrast examination shows normal location of the stomach, duodenal cap, and duodenojejunal (DJ) flexure to the left side of the spine and at the same level as the duodenal cap.
Figure 1: Fluoroscopic image of the upper gastrointestinal contrast examination shows normal location of the stomach, duodenal cap, and duodenojejunal (DJ) flexure to the left side of the spine and at the same level as the duodenal cap.

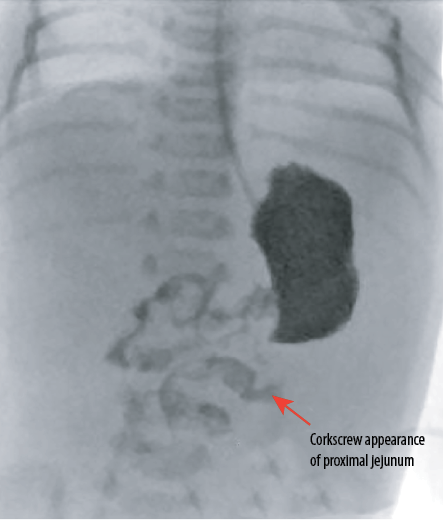 Figure 2: Fluoroscopic image of the upper gastrointestinal contrast examination obtained distal to duodenojejunal flexure shows coiling of the proximal jejunal loop giving a corkscrew appearance suggesting the possibility of volvulus.
Figure 2: Fluoroscopic image of the upper gastrointestinal contrast examination obtained distal to duodenojejunal flexure shows coiling of the proximal jejunal loop giving a corkscrew appearance suggesting the possibility of volvulus.
Upon exploratory laparotomy, MD was found with a peritoneal band extending from base of diverticulum to umbilicus, possibly causing intermittent volvulus and the consequent intestinal obstruction. A small cyst-like structure was noted in the middle of the band. No caliber change of bowel was noted at that level. The MD was resected, and the bowel underwent primary anastomosis. Feeds were started on the fourth postoperative day and increased quickly to full feeds. Karyotyping revealed T13, 47, XY + 13. The child kept tolerating feeds and went on to have an uneventful laparoscopy-assisted gastrostomy insertion. Pathology of the resected segment confirmed vitello-intestinal remnant on a small bowel segment in keeping with intraoperative findings. When correlated with the history of T13 and the clinical presentation, the contrast examination findings led us to conclude that the bowel obstruction was most likely due to symptomatic MD.
Discussion
T13 is one of the most common autosomal trisomies, accompanied by anomalies in several organs. The relation of T13 to a clinical syndrome was first recognized by Patau et al,2 in 1960 and it is also known as Patau’s syndrome. Median survival time for patients with T13 is only 7–10 days. Between 86% and 91% of live-born babies with T13 do not survive beyond the first year of life. Survival beyond the first year has been associated with mosaicism. A 19-year-old girl is the oldest known living person with regular T13.4
MD results from the formation of a true diverticulum in the small intestine as a result of an incomplete obliteration of the vitelline duct. It is often defined informally by the rule of twos: (1) occurs in approximately 2% of the population with a male-to-female ratio of 2:1, (2) is located within two feet from the ileocecal valve, (3) may manifest before the age of two years, and (4) is 2 inch long (though in practice variable).4,5 Although MD is a common congenital anomaly of the gastrointestinal tract, its symptomatic manifestation in the neonatal period is rare.6 Common presentations of neonatal MD that have been reported in the literature include bowel obstruction by inflammation, perforation, intussusception, segmental ileal dilatation, and ileal volvulus.7–11
The coexistence of T13 and symptomatic MD has been reported in only two cases based on our literature search,12,13 although there are scattered reports of such coexistence seen during autopsies of T13 patients. In this case, MD was discovered at the time of laparotomy for suspected volvulus with feed intolerance and bilious aspirates. MD has been diagnosed and reported earlier both during exploratory laparotomy for suspected volvulus/malrotation and more so as an incidental finding in a postmortem of T13 patients.6,13–18
Considering the rarity of symptomatic presentation of MD in T13 patients, it is not cost-appropriate to routinely recommend an isotope scan except perhaps in presence of suggestive GI symptoms. We think that even if the isotope scan (known to have a 30% false negative rate) is negative in a baby with features of volvulus/malrotation, there is still the need for urgent surgical exploration and correction.
In our case, a significant point of learning is reflected in the review of the upper GI contrast examination. The radiologist performing the upper GI contrast study should look for the proximal small bowel loops for their location, morphology, and contrast emptying, even though a clear demonstration of the normal duodenojejunal flexure is made. Otherwise, volvulus without malrotation can be missed.
The approach to the treatment of MD depends on whether it is discovered incidentally or as a result of symptoms. A physician may choose whether or not to surgically resect an incidentally discovered asymptomatic MD. However, a case of symptomatic MD must be treated by surgical resection, as in the current case.
Conclusion
Preoperative diagnosis of a symptomatic MD in a neonate with T13 is difficult, so it is necessary to maintain a high index of suspicion aided by delayed images of the contrast study. An early surgical intervention is recommended for a successful outcome. The possibility of MD should be considered in any child with T13 that presents with bowel obstruction, especially intermittent. Standard surgical treatment comprising MD resection and primary bowel anastomosis is generally safe and effective.
Disclosure
The authors declared no conflicts of interest. Consent was obtained from the patient’s parents.
references
- 1. Marcdante K, Kliegman R, Nelson W. Nelson Essentials of Pediatrics. 8th ed. Philadelphia, PA: Elsevier; 2019.
- 2. Edwards JH, Harnden DG, Cameron AH, Crosse VM, Wolff OH. A new trisomic syndrome. Lancet 1960 Apr;1(7128):787-790.
- 3. Passarge E, Stevenson RE. Meckel’s diverticulum. R.E. Stevenson, J.E. Hall (Eds.), Human malformations and related anomalies (2nd), Oxford University Press, Oxford (2006), p. 1111.
- 4. Redheendran R, Neu RL, Bannerman RM. Long survival in trisomy-13-syndrome: 21 cases including prolonged survival in two patients 11 and 19 years old. Am J Med Genet 1981;8(2):167-172.
- 5. Simms MH, Corkery JJ. Meckel’s diverticulum: its association with congenital malformation and the significance of atypical morphology. Br J Surg 1980 Mar;67(3):216-219.
- 6. Benson CD. Surgical implications of Meckel’s diverticulum. In: Ravicth MM, Welch KJ, Benson CD, eds. Pediatric Surgery. Chicago, IL: Year Book Medical Publishers; 1978:955-960.
- 7. Sy ED, Shan YS, Yang YR, Tsai HM, Lin CH. Hirschsprung’s disease, a rare precipitating factor in neonatal perforated Meckel’s diverticulum. J Pediatr Surg 2006 Jul;41(7):1319-1321.
- 8. Zahraa J, Abu-Ekteish F, Al Bassam AR, Nosir AA. Perforated Meckel’s diverticulum in a neonate mimicking necrotizing enterocolitis. Pediatr Emerg Care 2003 Dec;19(6):418-419.
- 9. Sy ED, Shan YS, Tsai HM, Lin CH. Meckel’s diverticulum associated with ileal volvulus in a neonate. Pediatr Surg Int 2002 Sep;18(5-6):529-531.
- 10. Ojha S, Menon P, Rao KL. Meckel’s diverticulum with segmental dilatation of the ileum: radiographic diagnosis in a neonate. Pediatr Radiol 2004 Aug;34(8):649-651.
- 11. Leconte D, Van Kote G, Questiaux E, Bader R, Charbonnel E, Khoury R. [A rare cause of neonatal occlusion by a palpable abdominal mass: Meckel’s diverticulum]. Chir Pediatr 1988;29(4):216-218.
- 12. Zúñiga S, Maira A, Zavala A. [Omphalocele, patent Meckel’s diverticulum and trisomy 13]. Rev Chil Pediatr 1986 May-Jun;57(3):270-272.
- 13. Alallah J, Al Talhi YM. Case 1: a full-term neonate with trisomy 13 and pneumoperitoneum. Neoreviews 2020 Aug;21(8):e571-e573.
- 14. Colacino SC, Pettersen JC. Analysis of the gross anatomical variations found in four cases of trisomy 13. Am J Med Genet 1978;2(1):31-50.
- 15. Aziz MA. Anatomical defects in a case of trisomy 13 with a D/D translocation. Teratology 1980 Oct;22(2):217-227.
- 16. Baty BJ, Blackburn BL, Carey JC. Natural history of trisomy 18 and trisomy 13: I. Growth, physical assessment, medical histories, survival, and recurrence risk. Am J Med Genet 1994 Jan;49(2):175-188.
- 17. Balci S, Güçer S, Orhan D, Karagöz T. A well-documented trisomy 13 case presenting with a number of common and uncommon features of the syndrome. Turk J Pediatr 2008 Nov-Dec;50(6):595-599.
- 18. Iijima S, Ohishi A, Mochida Y, Ohzeki T. Trisomy 13 and Meckel diverticulum: challenges in management of infants with trisomy 13. Am J Med Genet A 2007 Aug;143A(15):1749-1751.