Lipodystrophy is best defined as a heterogeneous group of syndromes manifested by variable loss of subcutaneous adipose tissue.1,2 Depending on the variability of fat loss, they can be classified into three groups; generalized, partial, and localized. The first two groups can be secondary to underlying genetic defects or acquired due to autoimmune mechanisms or drugs such as highly active antiretroviral therapy (HAART)-induced partial lipodystrophy.1 Based on these characteristics, four subgroups of generalized and partial forms are identified: congenital generalized lipodystrophy (CGL), acquired generalized lipodystrophy, familial partial lipodystrophy, and acquired partial lipodystrophy.3 Localized lipodystrophies, affecting small areas can result at sites of insulin and steroids injection.1 The localized and HIV-associated lipodystrophies are the most common while genetic and other acquired lipodystrophies are rare.1,4
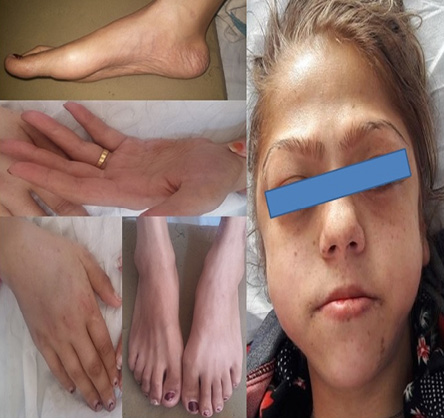
Figure 1: Acromegaloid features in our patient: prominent nose, supraorbital ridges and frontal bossing, large hands and feet, and preserved fat in cheeks, periorbital area, palm, and sole.
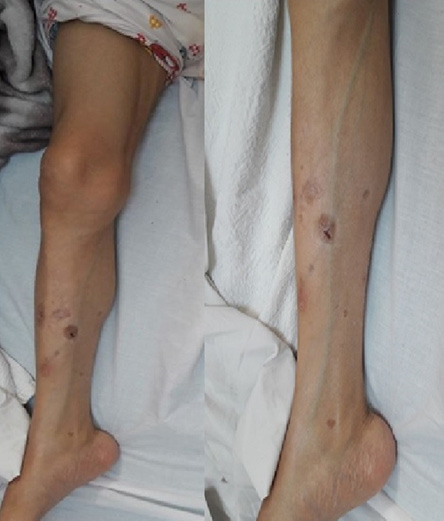
Figure 2: Prominent subcutaneous veins
and prominent thigh and calf muscles in a 30-year-old female.
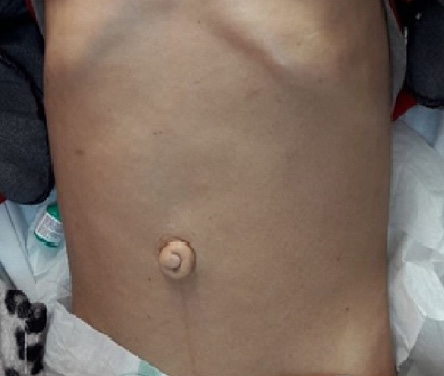
Figure 3: Prominent umbilicus in a 30-year-old female.
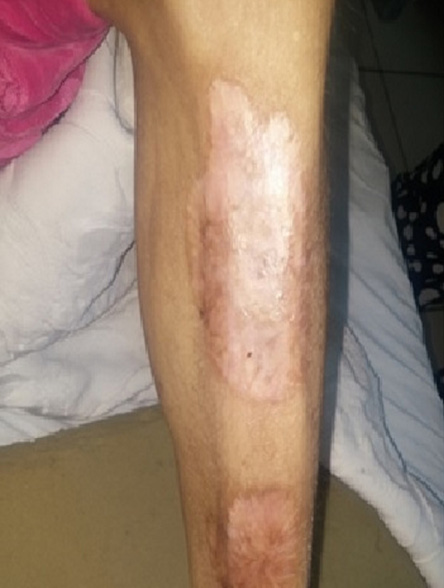
Figure 4: Shin lesions, which were confirmed to be necrobiosis lipoidica diabeticorum.
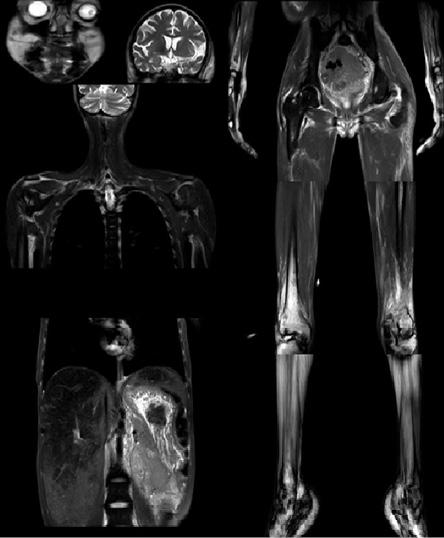
Figure 5: magnetic resonance imaging scan showing absent subcutaneous fat in the shoulder and pelvic girdles, arms, forearms, thighs, and legs. Also, there is preserved fat in the scalp, periorbital areas, cheeks, palms, and soles, in addition to hepatomegaly and fatty deposition in both liver and spleen.
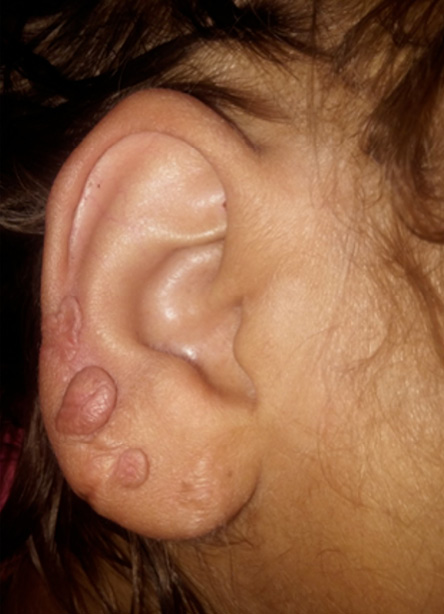
Figure 6: Keloid scars in the ear lobule and external auricles, which appeared at sites of healing of reactive perforating collagenosis lesions.
CGL or Berardinelli-Seip syndrome (BSS) is a rare autosomal recessive disorder.3,5 It has a prevalence of one in 10 million people.1 About 300 patients have been described in the literature as having generalized lipodystrophy.1,6,7 It is characterized by near to complete lack of subcutaneous fat starting at birth or infancy.5 As a consequence of absent functional adipose tissue, lipids are stored within other organs, including the liver, spleen, and skeletal and cardiac muscles. Accordingly, hepatomegaly and skeletal muscle hypertrophy are seen in virtually all cases.8,9
In early childhood, patients with CGL present with accelerated linear growth and advanced bone age, umbilical hernia or prominence, and acromegaloid features.2,3,5 Acanthosis nigricans, insulin resistance, ketosis-resistant diabetes mellitus (DM), and hypertriglyceridemia occur later in childhood or adolescence.1,3,5 Female patients may have polycystic ovary syndrome, irregular menstrual periods, hirsutism, clitoromegaly, and infertility.5,10 Male patients are reported to have normal reproductive function.10 Also, the absence of adipose tissue leads to a deficiency of important adipocytokines including leptin, resistin, and adiponectin.3,5 Leptin has an essential role in regulating glucose and lipid metabolism, an appetite suppressive effect, and an insulin-sensitizing effect.11,12 Accordingly, low leptin levels in patients with CGL lead to early childhood polyphagia, insulin resistance, and abnormal lipid metabolism, which further exacerbate the metabolic complications in these patients.2,5
Essential thrombocytosis (ET) is a myeloproliferative neoplasm (MPN) characterized by clonal proliferation of megakaryocytes leading to the uncontrolled production of platelets.13 It results from somatic mutations in JAK2V617F, calreticulin (CALR), and thrombopoietin receptor (MPL) genes in 70–90% of cases.14,15 In addition to thrombocytosis, patients may present with thrombosis or bleeding tendency, splenomegaly, leukocytosis, and leukemia transformation or progression to fibrosis.14,15 The first-line treatment of ET is hydroxyurea. Pegylated interferon-alpha, anagrelide, or busulfan are second-line treatments in patient who are intolerant or refractory to hydroxyurea.14 ET can be diagnosed based on the 2008 revised World Health Organization (WHO) diagnostic criteria, which requires the presence of all of the following: platelet count > 450 000/µL; megakaryocytic proliferation with little or no granulocytic or erythroid proliferation; not meeting the WHO criteria for chronic myeloid leukemia, polycythemia vera or other myeloid neoplasm; and demonstration of the JAK2V617F mutation, other clonal markers, or lack of evidence of a secondary cause.
We report here a rare case of BSS associated with ET with a complicated outcome; an unreported association to date.
Case report
A 30-year-old female, diagnosed with DM and hypertriglyceridemia at age 15, was referred to our clinic for management of poorly controlled DM. Before referral, she was assumed to have type 1 DM, and despite taking high doses of insulin, she was struggling for blood sugar control. She also had an irregular menstrual cycle, occurring 3–4 times/year, and hirsutism (Ferriman–Gallwey score of 10) in addition to thick and dry scalp hair.
At the time of initial evaluation, she was noted to have acromegaloid features with large hands and feet, prominent nose and supraorbital ridges, and frontal bossing [Figure 1]. She also has generalized lipoatrophy with no fat noted or felt over the trunk, buttocks, and limbs. Fat was preserved in her cheeks, palms, soles, and periorbital area [Figure 1]. Prominent muscles and superficial veins were also noted [Figure 2]. Her abdomen was protuberant along with inverted umbilicus, hepatomegaly, and splenomegaly [Figure 3]. She also had bilateral shin lesions with atrophied centers and red, elevated peripheries [Figure 4]. Her family confirmed that her current physical appearance was evident since infancy. They also reported that she had rapid height gain during childhood compared with her siblings and peers, and despite a voracious appetite, she did not gain weight and had an adult height similar to her relatives. Her parents are first degree cousins, and there was no family history of a similar condition.
The biochemical findings showed an increased liver enzymes concentration, hypertriglyceridemia with low high-density lipoprotein concentration, and high glycated hemoglobin (HbA1c) levels. Growth hormone, insulin-like growth factor, thyroid hormone levels, estradiol, luteinizing hormone, and follicle-stimulating hormone levels were normal. Mildly elevated testosterone levels was evident. Moreover, serological testing was negative for anti-glutamic acid decarboxylase 65 (anti-GAD 65) and anti-islets cell antibodies, ANA, and rheumatoid factor. Her creatine phosphokinase (CPK) levels, erythrocyte sedimentation rate, and C-reactive protein levels were all normal [Table 1]. Abdominal and ovarian ultrasound showed hepatomegaly with fatty infiltrates (30 cm span), splenomegaly (16 cm span), and polycystic ovaries. A whole-body magnetic resonance imaging (MRI) confirmed the presence of fat in the head, palms, and soles and loss of subcutaneous fat in other body parts [Figure 5]. Fatty deposition in grossly enlarged liver and in the spleen was also noted on MRI [Figure 5]. Furthermore, a biopsy from the shin lesions showed necrobiosis lipoidica diabeticorum.
She was managed with insulin, metformin, and anti-lipid agents (gemfibrozil and atorvastatin). During follow-up, due to a persistent high platelets count (756 000), which was first noticed on May 2013 and persisted for months [Table 2], in addition to high white blood cell count (16 500) and low hemoglobin (7 g/dL), a workup with blood film and bone marrow evaluation was done. A diagnosis of ET was made after ruling out infections, hemolysis, hematological cancers, iron deficiency anemia, and other inflammatory conditions, including rheumatoid arthritis and connective tissue diseases [Table 1]. JAK2V617F mutation was negative, testing for CALR and MPL genes mutations was not available. During further follow-up, the patient developed lower limb edema, and investigations revealed nephrotic range proteinuria (protein creatinine ratio of 4.1 mg/mg) with elevated creatinine (2.5 mg/dL). A kidney biopsy confirmed the presence of diabetic nephropathy. Her echocardiogram showed mild concentric left ventricular hypertrophy and an ejection fraction of 55%. Later, she developed pruritic nodular skin rash with central keratosis, and a skin biopsy revealed reactive perforating collagenosis. The rash healed with keloid scars in the ear lobules and external auricles [Figure 6]. The ET was managed first with hydroxyurea, and then she was prescribed weekly interferon injection with fair control of the disease [Table 2]. Unfortunately, a gradually deteriorating kidney function to stage V chronic kidney disease; manifesting with recurrent pulmonary edema and volume overload, required frequent admissions and commencement of renal replacement therapy by hemodialysis twice weekly.
Table 1: Laboratory results.
|
Aspartate transaminase |
45 U/L |
|
Alanine aminotransferase |
55 U/L |
|
Total bilirubin |
0.7 mg/dL |
|
HbA1c |
11.5% |
|
Triglyceride |
466 mg/dL |
|
Cholesterol |
195 mg/dL |
|
High-density lipoprotein |
32 mg/dL |
|
Low-density lipoprotein |
110 mg/dL |
|
Anti-nuclear antibodies |
Negative |
|
Rheumatoid factor |
Negative |
|
Insulin-like growth factor 1 |
204 ng/mL |
|
Estradiol |
65 pg/mL |
|
Testosterone |
55 ng/dL |
|
Luteinizing hormone |
2.4 uIU/mL |
|
Follicle-stimulating hormone |
3.5 uIU/mL |
|
Anti-glutamic acid decarboxylase and anti-islet cell antibodies |
Negative |
|
Creatine phosphokinase |
70 u/L |
|
Erythrocyte sedimentation rate |
25 mm/hr |
|
C-reactive protein |
Negative |
|
Ferritin |
35 ng/mL |
Discussion
BSS has at least four molecularly distinct forms resulting from mutation in one of the following genes: 1-acylglycerol-3- phosphate-O-acyltransferase 2 (AGPAT2) causing CGL1, Berardinelli-Seip congenital lipodystrophy type 2 (BSCL2) causing CGL2, caveolin-1 (CAV1) causing CGL3, and polymerase I and transcript release factor causing CGL4.1,2,10,16 These genes encode for different proteins, which are involved in the various steps of lipid droplet formation in adipocytes.9 AGPAT2 (CGL1) and BSCL2 (CGL2) mutations are the most common varieties accounting for about 95% of cases and have some overlapping and distinct phenotypic features.1,17 AGPAT2 gene is located on chromosome 9q34 and is an essential enzyme for triglyceride and phospholipid synthesis. It catalyzes the formation of phosphatidic acid from lysophosphatidic acid.1 The BSCL2 mutation encodes a transmembrane protein called seipin that is critical for the expression of key transcription factors involved in lipogenesis.9,18
Most of the complications observed in this syndrome are the consequence of adipose mass deficiency.10 Two types of white adipose tissue are described: functional and mechanical. Functional adipose tissue, which exerts metabolic activity, is missing in all CGL subtypes; while mechanical adipose tissue is maintained in CGL1, CGL3, and CGL4, but is absent in CGL2.1,10 In addition, CGL3 and CGL4 have preserved bone marrow fat, which is absent in patients with CGL1 and CGL2.1 In our case, the bone marrow fat was preserved as evidenced by bone marrow biopsy.
Patients with BSCL2 mutations have the most severe variety of CGL,1 they present with more severe and premature metabolic manifestations,19 in addition to a higher incidence of intellectual deficiency (80%) and cardiomyopathy.10 This is because seipin is expressed variably in several tissues and is highly expressed in the central nervous system.9,10,18,20 Also, liver failure and a more generalized absence of adipose tissue involving the palms, soles, scalp, and periorbital regions correspond more to CGL2.3,10,18 CGL1 has a less severe presentation compared to CGL2 and has been accompanied by sclerotic bone lesions before and lytic bone lesions after puberty.10 CGL3 patients, in addition to metabolic complications, also present with short stature and vitamin D resistance.1 CGL4 patients suffer from congenital myopathy, percussion-induced myoedema, atlantoaxial instability, and cardiac rhythm disturbances.1
Table 2: Serial complete blood count results.
|
White blood cell |
16.500 |
15.600 |
17.600 |
13.400 |
11.200 |
8.600 |
7.100 |
6.800 |
|
Red blood cell, mcL |
3.2 |
3.3 |
3.25 |
3.8 |
4.1 |
5.1 |
5.2 |
5.5 |
|
Hemoglobin, g/dL |
7.1 |
7.5 |
7.3 |
8.1 |
8.5 |
11.6 |
11.7 |
12.1 |
|
Hematocrit, % |
22.0 |
21.0 |
24.0 |
27.0 |
28.5 |
33.0 |
34.0 |
35.0 |
ET: essential thrombocytosis.
Due to its characteristic features, BSS is usually diagnosed at birth or within the first year of life. Although in some patients, as our case, it might be diagnosed later in adulthood.3,10 Most of CGL features were positive in our case, and as the features were evident since infancy, a clinical diagnosis of BSS was made.
Thrombocytosis can be primary (also known as ET) or secondary to infections, hemolysis, iron deficiency anemia, asplenia, and inflammatory diseases as rheumatoid arthritis.21 The association of BSS with ET was not reported before to the best of our knowledge. This association can be a coincidence or might be related to an unrecognized genetic predisposition for both diseases. Unfortunately, genetic testing was not done as it is not available in our institution. In addition, the presence of the skin manifestations, including reactive perforating collagenosis and necrobiosis lipoidica diabeticorum, makes this case unique. Although a genetic study was not done, the absence of intellectual impairment, the preserved fat in the head, palms and sole, and the absence of other features seen in CGL2, CGL3, and CGL4, suggest the diagnosis of AGTPA2 mutation (CGL1) as the most likely.
The treatment of CGL can be difficult, but lifestyle modifications and conventional antihyperglycemic and lipid-lowering medications are usually used.3,7 Insulin, sulfonylureas, metformin, and pioglitazone are used to manage hyperglycemia, while fibrates and statins can be used to manage hypertriglyceridemia. Plasmapheresis is an option for lowering dangerously high triglyceride levels to control painful xanthomas and prevent pancreatitis.3 In our case, the metabolic derangement was controlled with dietary measures, insulin, and anti-lipid agents. Metformin was also used, but was stopped after she developed renal impairment. However, in severe cases, these measures may fail to control metabolic derangement. Leptin therapy can be beneficial in such conditions, especially in severe insulin resistance and steatohepatitis. Recombinant leptin such as metreleptin has been approved as a therapeutic agent by the Food and Drug Administration since February 2014.3,6,22
Conclusion
CGL is usually diagnosed during infancy, but being a rare disease, the medical personnel may be unfamiliar with it, leading to late diagnosis. We present a rare case of CGL, which was diagnosed at the age of 30 years. The features of the patient are mostly consistent with AGTPA2 mutation (CGL1). A previously non-reported association with ET was also observed. In addition, skin biopsies from two different lesions were consistent with reactive perforating collagenosis and necrobiosis lipoidica diabeticorum. The patient metabolic complications were controlled on conventional treatment with insulin and anti-lipids agents and regular hemodialysis.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Garg A. Clinical review: lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011 Nov;96(11):3313-3325.
- 2. Huang-Doran I, Sleigh A, Rochford JJ, O’Rahilly S, Savage DB. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol 2010 Dec;207(3):245-255.
- 3. Handelsman Y, Oral EA, Bloomgarden ZT, Brown R, Chan J, Einhorn D, et al. The clinical approach to the detection of lipodystrophy–an AACE consensus statement. Endocrine Practice 2013;19(1):107-116.
- 4. Fiorenza CG, Chou SH, Mantzoros CS. Lipodystrophy: pathophysiology and advances in treatment. Nat Rev Endocrinol 2011 Mar;7(3):137-150.
- 5. Hussain I, Abhimanyu G. Lipodystrophy syndromes. Dermatologic Clin 2008;26(4):569-ix.
- 6. Simha V. Metreleptin for metabolic disorders associated with generalized or partial lipodystrophy. Expert Rev Endocrinol Metab 2014 May;9(3):205-212.
- 7. Kazandjieva J, Guleva D, Marina S, Nikolova A, Mladenova G, Kurtev A. Berardinelli-seip syndrome - a case report. Serbian Journal of Dermatology and Venereology 2016;8(2):101-104.
- 8. Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, et al. The diagnosis and management of lipodystrophy syndromes: a multi-society practice guideline. J Clin Endocrinol Metab 2016 Dec;101(12):4500-4511.
- 9. Purizaca-Rosillo N, Mori T, Benites-Cóndor Y, Hisama FM, Martin GM, Oshima J. High incidence of BSCL2 intragenic recombinational mutation in Peruvian type 2 Berardinelli-Seip syndrome. Am J Med Genet A 2017 Feb;173(2):471-478.
- 10. Hasani-Ranjbar S, Soltani A, Hadavi M, Ejtahed HS, Mohammad-Amoli M, Radmard AR. Congenital generalized lipodystrophy in a youth presented with sclerotic and lytic bone lesions; a family with AGPAT2 mutation. Int J Pediatr 2017;5(2):4275-4284.
- 11. Park H-K, Ahima RS. Physiology of leptin: energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015 Jan;64(1):24-34.
- 12. Eteiwi S, Al-Asaad RA, Aleyadeh AA, Al-Sarihin KK, Alzu’bi AA, Obeidat AA, et al. Leptin as a predictor of cardiovascular disease: A cross- sectional study in a sample of Jordanian patients. Journal of the Royal Medical Services 2017;102(5512):1-9.
- 13. Ayres-Silva JP, Bonamino MH, Gouveia ME, Monte-Mor BC, Coutinho DF, Daumas AH, et al. Genetic alterations in essential thrombocythemia progression to acute myeloid leukemia: a case series and review of the literature. Frontiers in Oncology 2018;8:32.
- 14. Tefferi A, Vannucchi AM, Barbui T. Essential thrombocythemia treatment algorithm. Blood Cancer Journal 2018;8(1):1-6.
- 15. Zaidi U, Shahid S, Fatima N, Ahmed S, Sufaida G, Nadeem M, et al. Genomic profile of a patient with triple negative essential thrombocythemia, unresponsive to therapy: a case report and literature review. J Adv Res 2017 Jul;8(4):375-378.
- 16. Ramanathan N, Ahmed M, Raffan E, Stewart CL, O’Rahilly S, Semple RK, et al. Identification and characterisation of a novel pathogenic mutation in the human lipodystrophy gene AGPAT2 : C48R: a novel mutation in AGPAT2. JIMD Rep 2013;9:73-80.
- 17. Shastry S, Delgado MR, Dirik E, Turkmen M, Agarwal AK, Garg A. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A 2010 Sep;152A(9):2245-2253.
- 18. Gonzalo MM, Estefania CV. Congenital generalized lipodystrophy type 2 in a patient from a high-prevalence area. J Endocr Soc 2017 Jun;1(8):1012-1014.
- 19. Hegele RA, Joy TR, Al-Attar SA, Rutt BK. Thematic review series: adipocyte biology. Lipodystrophies: windows on adipose biology and metabolism. J Lipid Res 2007 Jul;48(7):1433-1444.
- 20. Van Maldergem L, Magré J, Khallouf TE, Gedde-Dahl T Jr, Delépine M, Trygstad O, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet 2002 Oct;39(10):722-733.
- 21. Rose SR, Petersen NJ, Gardner TJ, Hamill RJ, Trautner BW. Etiology of thrombocytosis in a general medicine population: analysis of 801 cases with emphasis on infectious causes. J Clin Med Res 2012 Dec;4(6):415-423.
- 22. Rother KI, Brown RJ. Novel forms of lipodystrophy: why should we care? Diabetes Care 2013 Aug;36(8):2142-2145.