Eosinophilic granulomatosis with polyangiitis (EGPA) is a rare vasculitis characterized by necrotizing granulomatous inflammation of the respiratory tract and necrotizing vasculitis of the small to medium vessels. EGPA associated with anti-neutrophil cytoplasmic antibodies (ANCAs) positivity, especially in the presence of glomerulonephritis, was first reported in the 2012 revised International Chapel Hill Consensus Conference.1 ANCA, however, is less frequently associated with EGPA compared to other vasculitis, especially in children.2 Classically, patients present with respiratory symptoms (e.g., asthma), rhinitis, pulmonary infiltrates, neuropathy, or evidence of extravascular eosinophils with peripheral eosinophilia (> 10% of total white cell count).3,4 Cardiac and renal involvement as well as the duration of the disease, may be associated with a bad outcome. EGPA typically has a male preponderance with an age range of 20 to 50 years old. Childhood-onset has been reported rarely, and cutaneous manifestation is uncommon.5
Case report
A 15-year-old male with no comorbidities complained of multiple itchy papules on both of legs for six months associated with a painful indurated plaque for two months. The lesion started as itchy papules on both legs six months prior for which he did not seek treatment. He started to develop painful indurated plaques on both legs associated with non-pitting edema four months after the onset of itchy papules. He denied any insect bite or injury before the onset of the skin lesions. There was no fever, cough, wheezing, shortness of breath, arthralgia, loss of appetite, or weight loss. He had neither asthmatic symptoms nor rhinitis throughout his childhood. There was no known history of atopy or allergy and no family history of any similar problem.
Clinically, he was afebrile with normal vital signs. There were multiple excoriated erythematous papules on both legs with indurated tender plaques on both shins [Figure 1]. He had bilateral inguinal lymphadenopathy (3 cm × 2 cm), but no other peripheral lymphadenopathy. There was no hepatosplenomegaly and no significant cardiovascular, respiratory, or neurological findings. Head and neck examinations were unremarkable.
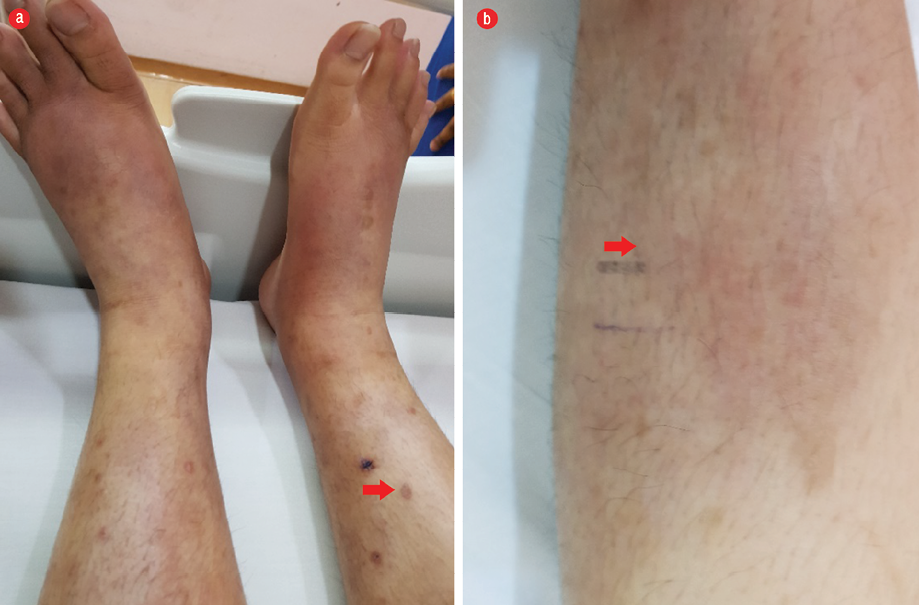
Figure 1: Lower limbs of the patient showing (a) multiple excoriated erythematous papular rash (red arrow) and (b) indurated tender non-pitting plaque (red arrow).
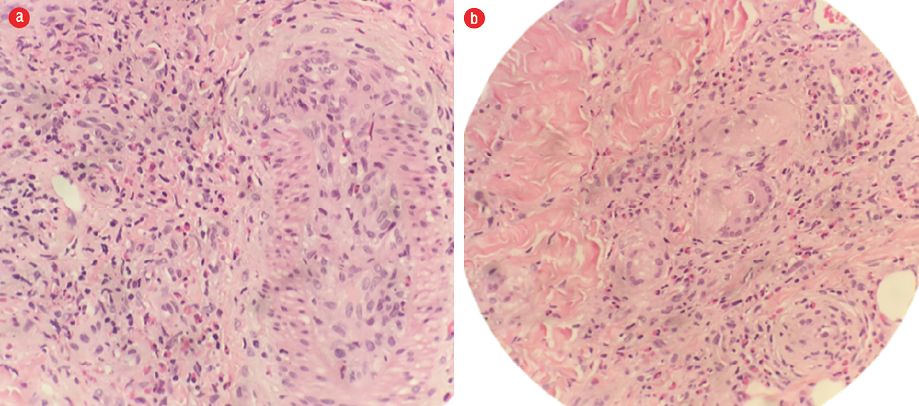
Figure 2: Hematoxylin and eosin staining of skin biopsy shows (a) prominent eosinophilic infiltration and eosinophilic angiitis and (b) granuloma formation with aggregates of multinucleated giant cells. Magnification = 40 ×.
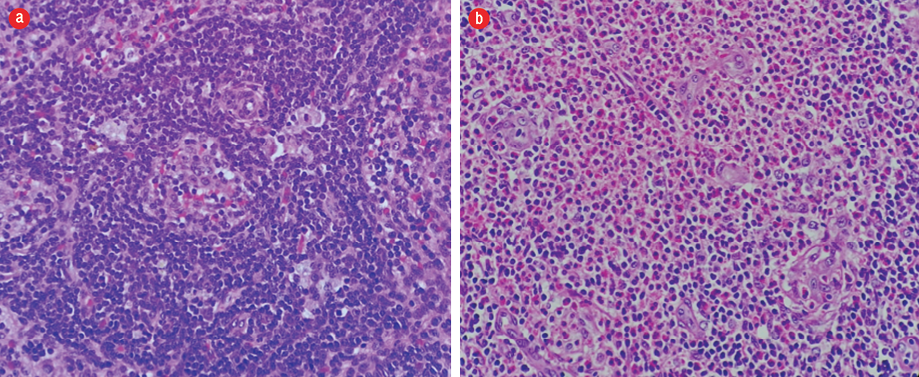
Figure 3: Hematoxylin and eosin staining of histopathology from inguinal lymph node biopsy showing (a) dilated sinuses containing histiocytes, lymphocytes, and occasional pigment-containing histiocytes and (b) eosinophils infiltration, forming microabscesses. Magnification = 400 ×.
His complete blood count (performed three months prior to this visit) was normal with a white cell count (WCC) of 12.2 × 109/L (4.5 – 13.0 × 109/L), and normal absolute eosinophil count (AEC) of 244 cells/µL, which gradually increased over the next three months to 13 688 cells/µL (WCC of 23.6 × 109/L). His WCC showed an increasing trend, with normal counts of neutrophils and platelets, and a normal hemoglobin level. His serum creatinine level was within the normal range. Immunological markers were negative (anti-nuclear antibody, complement 3 and 4, rheumatoid factor, and cytoplasmic and perinuclear ANCA). Coombs test was negative, and glucose-6-phosphate dehydrogenase was normal. Immunoglobulin E (IgE) level was markedly raised at 1108 IU/mL (< 100 IU/mL). The liver profile, including alkaline phosphatase, was within normal range. Globulin was normal, and lactate dehydrogenase levels were mildly raised at 246 U/L (135–225 U/L). His erythrocyte sedimentation rate was 18 mm/hr. Peripheral blood film showed leukocytosis and eosinophilia but no blast cells. Hepatitis and retroviral screening, venereal disease research laboratory test, blood culture, stool ova, and cyst tests were negative. His creatinine kinase, creatine kinase-muscle/brain, high sensitivity troponin T, and coagulation profile were also normal. Chest radiograph and ultrasonography of the abdomen were normal.
Skin biopsies taken from the lower limb showed blood vessels in the deeper dermis with a thickened wall infiltrated by lymphocytes and eosinophils. However, there was no fibrinoid necrosis seen. The dermis and the subcutaneous tissue septa and part of the lobules were densely infiltrated by eosinophils admixed with lymphocytes and histiocytes forming flame bodies. Scattered granuloma formation composed of a collection of epithelioid histiocytes associated with multinucleated giant cells were present. We saw no evidence of malignancy [Figure 2]. Meanwhile, the lymph node biopsies showed features consistent with dermatopathic lymphadenopathy with normal immunoarchitecture (CD20, BCL2, Ki67, CD3, and CD5) [Figure 3]. The histopathological findings were in favor of EGPA.
He was given oral prednisolone 40 mg and azathioprine 50 mg daily. His AEC dropped to 19% 48 hours after initiation of prednisolone with resolution of the edema. His WCC also gradually decreased over time. There was no systemic symptom otherwise. At eight weeks, his full blood count was normal (AEC = 696 cells/µL). The skin lesions had completely resolved.
Discussion
EGPA (formerly known as Churg-Strauss syndrome) was first described by Jacob Churg and Lotte Strauss in 1951 and was considered as one of the rarest systemic vasculitis affecting the medium and small caliber vessels.3 It is a new terminology introduced in 2012 revised International Chapel Hill Concensus Conference nomenclature of vasculitides. The criteria for diagnosis requires presence of four or more of the six features mentioned previously.3,4 It has been commonly reported in adults with age of onset between 25 and 50 years old with significant morbidity and mortality. It is rare in childhood.6–8
Asthma has been well recognized as the cardinal prodromal symptom. This may be present long before other systems are involved. Classically, this is followed by peripheral eosinophilia and systemic vasculitis in the third phase of the condition. Cutaneous manifestations have been commonly observed to co-exist with other systemic involvement, such as pulmonary, cardiac, neurological, and gastrointestinal systems. ANCAs are positive in less than 40% EGPA cases compared with other ANCA-associated vasculitides.2,9
Cutaneous manifestation in EGPA without other systemic involvement has yet to be reported in the literature or case series of both adults and children. Hence, it was a great challenge to reach the diagnosis of EGPA in this young boy. The signs and symptoms of this patient could have been due to several other conditions, some of which may be life-threatening. The presence of cutaneous vasculitis over the lower limbs, significant peripheral hypereosinophilia, and raised IgE may occur in allergic disorders, for example, IgG4-related disease, pulmonary eosinophilia, drug hypersensitivity, parasitic infections, malignancies, hypereosinophilic syndrome, and other inflammatory disorders (e.g., granulomatous with polyangiitis and microscopic polyangiitis), which have distinct clinical and laboratory manifestations.9 Histopathological findings of predominantly eosinophilic inflammation and necrotizing vasculitis with granulomatous changes affecting the small and medium caliber vessels in EGPA were very useful to differentiate from other forms of vasculitis. There are cases of EGPA reported in childhood presenting with a various spectrum of systemic involvement and cutaneous vasculitis occurring as an early presentation.5,10–12 Kimura’s disease may present with similar findings of marked peripheral blood eosinophilia, elevated serum IgE, and histopathological findings of the hyperplastic lymphoid follicle with eosinophilic infiltration and microabscesses, fibrocollagenous deposition, and vascular proliferation.13,14 However, the clinical features of Kimura’s disease described in previous case series were not present in our patient,13,15 and the presence of granulomatous angiitis from the skin biopsy make Kimura’s less likely to be the etiology. The presence of pigment-containing histiocytes in the lymph node biopsy favors dermatopathic lymphadenopathy rather than Kimura’s disease.
Corticosteroid remains the mainstay of treatment followed by maintenance therapy with immunosuppressants such as azathioprine, methotrexate, and cyclophosphamide. However, this is recommended in patients with EGPA who have systemic involvement or life-threatening manifestations to induce remission and prevent relapse.16 Biologic agents have not been used in children in previous case series, and there is limited data in adult studies, although it looks promising.17,18
Conclusion
Though it is rare in childhood, cutaneous vasculitis and peripheral eosinophilia with classical histopathological findings may represent a distinctive early clinical feature of EGPA without other major organ or systemic involvement and should be strongly considered. Treatment should be started as soon as the diagnosis is made to prevent further progression of this potentially life-threatening disease.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum 2013 Jan;65(1):1-11.
- 2. Zwerina J, Eger G, Englbrecht M, Manger B, Schett G. Churg-Strauss syndrome in childhood: a systematic literature review and clinical comparison with adult patients. Semin Arthritis Rheum 2009 Oct;39(2):108-115.
- 3. Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) 1984 Mar;63(2):65-81.
- 4. Mouthon L, Dunogue B, Guillevin L. Diagnosis and classification of eosinophilic granulomatosis with polyangiitis (formerly named Churg-Strauss syndrome). J Autoimmun 2014 Feb-Mar;48-49:99-103.
- 5. Sahin S, Adrovic A, Barut K, Kasapcopur O. Childhood-onset eosinophilic granulomatosis with polyangiitis: a rare childhood vasculitis mimicking anthrax and eosinophilic leukaemia. BMJ Case Rep 2016 Feb;2016:bcr2015213856.
- 6. Boyer D, Vargas SO, Slattery D, Rivera-Sanchez YM, Colin AA. Churg-Strauss syndrome in children: a clinical and pathologic review. Pediatrics 2006 Sep;118(3):e914-e920.
- 7. Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al; French Vasculitis Study Group. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum 2013 Jan;65(1):270-281.
- 8. Eleftheriou D, Gale H, Pilkington C, Fenton M, Sebire NJ, Brogan PA. Eosinophilic granulomatosis with polyangiitis in childhood: retrospective experience from a tertiary referral centre in the UK. Rheumatology (Oxford) 2016 Jul;55(7):1263-1272.
- 9. Nutman TB. Evaluation and differential diagnosis of marked, persistent eosinophilia. Immunol Allergy Clin North Am 2007 Aug;27(3):529-549.
- 10. Rodríguez-Armendáriz R, Hernández-Saldaña R, Hinojos-Gallardo LC, Ramos-Martínez E, Soto-Ramos M. Granulomatosis eosinofílica con poliangitis: reporte de un caso y revisión de la literatura. Neumol Cir Torax 2017;76(1):36-43.
- 11. Yüksel S, Evrengül H, Becerir T, Koçyiğit A, Cinbiş M, Demirkan N. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) without respiratory symptoms in a boy. Pediatr Rheumatol Online J 2014;12(Suppl 1):366.
- 12. El-Gamal Y. Churg-Strauss syndrome in the pediatric age group. World Allergy Organ J 2008 Feb;1(2):34-40.
- 13. Sia KJ, Kong CK, Tan TY, Tang IP. Kimura’s disease: diagnostic challenge and treatment modalities. Med J Malaysia 2014 Dec;69(6):281-283.
- 14. Chen H, Thompson LD, Aguilera NS, Abbondanzo SL. Kimura disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 2004 Apr;28(4):505-513.
- 15. Chokkappan K, Al-Riyami AM, Krishnan V, Min VL. Unusual cause of swelling in the upper limb: kimura disease. Oman Med J 2015 Sep;30(5):372-377.
- 16. Groh M, Pagnoux C, Baldini C, Bel E, Bottero P, Cottin V, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med 2015 Sep;26(7):545-553.
- 17. Mohammad AJ, Hot A, Arndt F, Moosig F, Guerry MJ, Amudala N, et al. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Ann Rheum Dis 2016 Feb;75(2):396-401.
- 18. Jachiet M, Samson M, Cottin V, Kahn JE, Le Guenno G, Bonniaud P, et al; French Vasculitis Study Group. Anti-IgE monoclonal antibody (omalizumab) in refractory and relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss): data on seventeen patients. Arthritis Rheumatol 2016 Sep;68(9):2274-2282.