| |
Abstract
Thrombocytopenia is a well- recognized complication of heparin therapy. The diagnosis is mostly clinical and the main value of laboratory testing is in excluding the diagnosis. We describe here a patient with stroke who had aspiration pneumonia leading to sepsis. She developed atrial fibrillation and received heparin which had to be stopped prematurely due to melena. Within 5 days of heparin, she had thrombocytopenia which was heparin-induced thrombocytopenia (HIT), but the diagnosis was missed initially as heparin was no longer on the drug chart.
Keywords: Heparin-induced thrombocytopenia; Atrial fibrillation; Stroke.
Introduction
Thrombocytopenia is a well-recognized complication of heparin therapy, usually occurring within 5 to 10 days after heparin treatment.1 There are two types; type I and type II. Type I heparin-induced thrombocytopenia which appears to be of no clinical consequence and is due to a non-immune mediated direct effect of heparin on platelet activation.2 It is typically characterized by a lesser fall in platelet count that occurs within the first two days after heparin initiation and often returns to normal with continued heparin administration. On the other hand, type II heparin-induced thrombocytopenia (HIT), a more serious form, is an immune-mediated disorder characterized by the formation of IgG antibodies against the heparin-platelet factor IV complex. The antibodies then bind to platelets, which activates them. Platelet activation leads to both thrombocytopenia and a pro-thrombotic state. The platelet drop is more severe and usually occurs 5 to 10 days after start of heparin. For the remainder of this review, the term heparin-induced thrombocytopenia (HIT) will refer only to the immune form (i.e., type II), while HITT stands for heparin-induced thrombocytopenia and thrombosis.
This report describes a case of HIT in a patient with stroke complicated by aspiration pneumonia, sepsis syndrome, aortic stenosis, and atrial fibrillation, who developed both thrombocytopenia and arterial and venous thromboses. HIT is not a very uncommon disorder but needs a high index of clinical suspicion as the diagnosis is mostly clinical and laboratory confirmation is either not available, as in our case, or is delayed. The diagnosis may not be as straight forward as it appears to be, especially in a critically ill patient who may have several other causes of reduced platelet counts like disseminated intravascular coagulation (DIC), drug or transfusion induced thrombocytopenia. We present this case to highlight its importance and to create an increased awareness of this serious and potentially fatal disorder.
Case Report
An 83-year-old Omani female was admitted to Sultan Qaboos Hospital in February 2011 due to left-sided hemiplegia of about 12-hour duration which had started in the early morning. She had been bedridden for 3 months because of osteoarthritis particularly affecting the knees. She was reported to be suffering from peptic ulcer disease diagnosed on endoscopy about 3 months ago, but she was not known to have hypertension or diabetes. On examination she was confused; blood pressure 122/84 mm Hg, pulse 108/min, temperature 37.3°C, and oxygen saturation on room air was 100% by pulse oximetry. She had slurred speech, left facial palsy of upper motor neuron type, and left sided hemiplegia with power grade 1/5 in left leg and 0/5 in left arm. Left plantar was upgoing. Urgent computerized tomographic scan (CT scan) of the brain did not reveal any hemorrhage or infarct, and repeat CT brain on the 5th day of admission was unchanged. The investigations showed white cell count 7.6 × 109/L, hemoglobin 11 g/dL, platelets 460 × 109/L, international normalized ratio (INR) 0.94, activated partial thromboplastin time (APTT) 28.5 sec, blood sugar 5.53 mmol/L, normal liver enzymes, electrolytes, and creatinine.
She was managed as suffering from acute stroke with probable brain infarction and started on aspirin 75 mg per day and dipyridamole 75 mg three times daily. On day 4, she was febrile and slight swelling of left leg was noticed. Doppler study of the legs was carried out, but it did not show any thrombi. Ceftazidime was added for presumed chest infection. Next day (day 5) her condition was quite serious with dyspnea, hypotension (blood pressure 90/60 mmHg), and hypoxemia. Oxygen saturation was being maintained up to 96% with high flow oxygen and nebulizations. Chest X-ray showed left basal pneumonia. Intravenous fluids were pushed, dopamine and low dose steroids were started, and injectable metronidazole was added for aspiration pneumonia.
She continued to remain in critical condition. On day 9, she developed atrial fibrillation and oral digoxin and heparin infusion were started. Heparin had to be stopped next day as she had melena. Aspirin and dipyridamole were also stopped. She was already on oral omeprazole. Her platelet count on day 9, when heparin was started, was 462 × 109/L. Next day (on day 10), a bed-side echocardiogram was done because of atrial fibrillation, and calcific aortic stenosis with a gradient of 45 mmHg was discovered. Left ventricular systolic function was found to be fair (ejection fraction 45%).
She received multiple courses of antibiotics including meropenem, tazocin, and amikacin, but remained in need of inotropes to maintain the blood pressure. The liver and kidney function tests were normal. Blood and urine cultures sent earlier on day 4, and repeat blood cultures on day 18, turned out to be sterile. On day 24, ischemic discoloration was noted on the left forefoot. The Doppler study revealed monophasic low velocity blood flow in the posterior tibial and dorsalis pedis arteries on the left. There were venous thrombi in both lower limbs including the femoral veins, being more extensive on the left side. Thrombus was also noted in right internal jugular vein extending to the superior vena cava on Doppler study. CT scan of the chest with pulmonary angiogram was also carried out and it showed thrombus in the right internal jugular vein, superior vena cava and partial thrombosis in right and left branches of pulmonary artery. (Figs. 1-3)
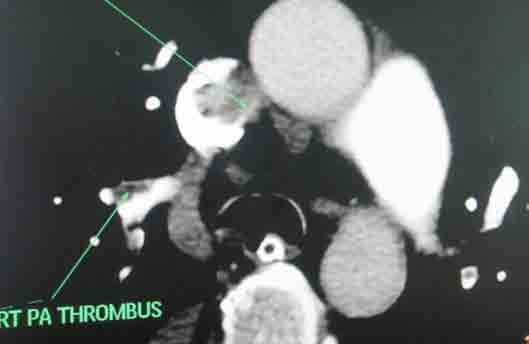
Figure 1: Axial section of CT scan with pulmonary angiogram showing thrombosis in superior vena cava and right pulmonary artery.
A consolidation was noted in right lower lobe. She needed anticoagulation, but because of prior melena, upper endoscopy was carried out on day 25. It showed a large hiatus hernia with distal esophagitis, multiple areas of submucosal bleeding spots and superficial ulcers in stomach. These lesions were assessed to be at low risk for bleeding, and Fondaparinux 2.5 mg subcutaneously daily was started. Platelet counts are shown in Fig. 4. It is clear that although she received unfractionated heparin infusion only for one day, on day 9, when platelets were 462 × 109/L, the platelets started to have a progressive downward trend beginning on day 14 (5th day after initiation of heparin), falling to 303 × 109/L on day 15, and were 135 × 109/L on day 18 (9th day after heparin), which is less than 50% of the counts on initiation of heparin (from 462 to 135 × 109/L). The low platelet count was confirmed by a peripheral blood smear.
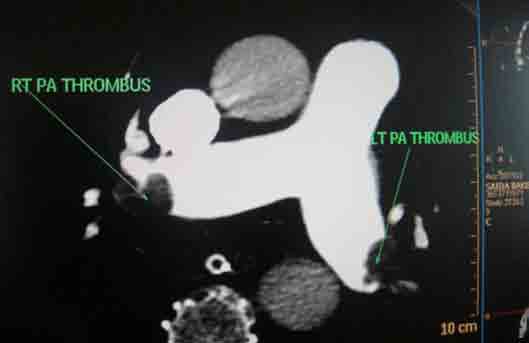
Figure 2: Axial section of CT scan with pulmonary angiogram showing main pulmonary artery with thrombi in the left and the right branches.
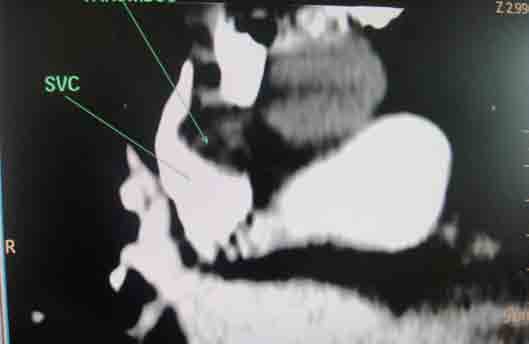
Figure 3: Coronal section of CT scan with pulmonary angiogram showing thrombosis in superior vena cava.
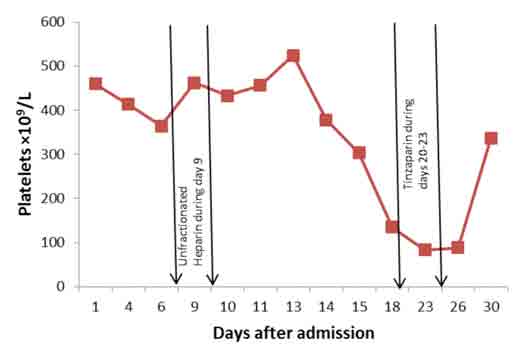
Figure 4: Platelet counts on different days after admission with periods of heparin administration between the arrows.
The diagnosis of heparin as a cause of thrombocytopenia (HIT) was missed at this point as heparin was no longer on the drug chart. Actually, she also happened to receive tinzaparin, a low molecular weight (LMW) heparin, during days 20 to 23 as prophylaxis for deep vein thrombosis (DVT). Platelets dropped further and reached a nadir of 83.1 × 109/L on day 23. Platelets remained as low as 87.8 /L until day 26. Presence of extensive thromboses, both arterial and venous, in spite of thrombocytopenia suggested the diagnosis of HIT on day 24. Prothrombin time (PT) and activated partial thromboplastin time (APTT) remained within normal range during these days, except for transiently prolonged APTT on day 10 secondary to heparin. There was no evidence of DIC or any other explanation for thrombocytopenia. Fondaparinux 2.5 mg subcutaneously daily was started on day 25 for presumed HIT. Laboratory confirmation could not be done as the HIT (anti-heparin/PF4) antibodies were not available. Her clinical condition gradually improved slightly and platelet count rose to 336 × 109/L by day 30. She was doing well but on day 33, she vomited and unfortunately aspirated. She desaturated, went into respiratory failure and refractory shock and died on the same day.
Discussion
HIT occurs with a frequency of 0.2% to 5.0% in patients exposed to unfractionated heparin (UFH) for more than four days, with an overall incidence of 2.6% noted in a meta-analysis.3 The incidence is closer to 0.2% for those treated with unfractionated heparin for less than four days.4 The incidence appears to be lower with LMW heparin.5 HIT is more common in surgical patients and in females.5
HIT results from antibody formation provoked not by heparin alone but by a complex of heparin and the platelet-specific protein platelet factor 4 (PF4), a heparin-neutralizing protein contained in the alpha granules of platelets, which is released from the platelet upon its activation.6 The anti-heparin/PF4 antibody can activate platelets through the Fc gamma RIIa receptor and also likely activates endothelial cells, leading to additional platelet activation with further release of PF4, creating a positive feedback loop.7 Activated platelets with the heparin/PF4 antibody complex attached to their surface undergo aggregation, and are removed prematurely from the circulation leading to thrombocytopenia (HIT), and the generation of procoagulant platelet-derived microparticles, frequently resulting in thrombin generation and thrombosis (HITT). Many patients exposed to heparin develop antibodies to heparin/PF4, but do not appear to have adverse consequences. A fraction of those who develop antibodies will develop thrombocytopenia, and a portion of those (up to 50%) will develop HIT and thrombosis (HITT).
Drug-induced thrombocytopenia due to heparin differs from that seen with other drugs in two major ways. The thrombocytopenia is not usually severe, being on average 60 × 109/L, with nadir counts rarely <20 × 109/L; and secondly the HIT is not associated with bleeding; and in fact, markedly increases the risk of thrombosis, both venous and arterial, which can be detected in about 50% of the patients. Venous thrombosis can be complicated by pulmonary embolism, the most common life-threatening event, venous limb gangrene (distal ischemic necrosis following deep vein thrombosis), and cerebral sinus thrombosis. Arterial thrombosis, although less common, can lead to stroke, myocardial infarction, acute limb ischemia from peripheral arterial occlusion, or organ infarction (mesentery, kidney).
Platelet counts need to be monitored frequently during heparin administration. Immune-mediated HIT is associated with a fall in the platelet count of >50% that typically occurs 5 to 10 days after the initiation of heparin therapy. Onset after two weeks is unusual. It may occur before 5 days in those who were exposed to heparin in the prior few weeks or months (<100 days) and have circulating anti-heparin/PF4 antibodies.8 Rarely, thrombocytopenia and thrombosis begin several days after all heparin has been stopped (termed delayed onset HIT).9
HIT antibodies (anti-heparin/PF4 antibodies) can be detected using two types of assays. The most widely available is an enzyme-linked immunoassay (ELISA). This is a very sensitive assay (91% to >97%); a negative test strongly suggests the absence of HIT, but the test has a low specificity for the diagnosis of HIT since many patients develop antibodies but do not develop clinical HIT. The other assay is a functional platelet activation assay that measures the ability of the patients' serum to activate platelets in the presence of heparin, like serotonin release assay (sensitivity and specificity >95% but high cost) or platelet aggregation assay (specificity >90% but suffers from lack of sensitivity). However, HIT remains a clinical diagnosis. The main value in testing is in excluding the diagnosis with negative tests, particularly ELISA. The 4 T’s have been recommended to be used in a diagnostic algorithm for HIT: thrombocytopenia, timing of platelet count drop, thrombosis and other sequelae such as localized skin reactions, and other cause of thrombocytopenia not evident.10
Treatment should be initiated as soon as the diagnosis of HIT is suspected before results of laboratory testing is available. Exposure to all forms of heparin should be immediately discontinued, including heparin-bonded catheters and heparin flushes.11 Because of the high rate of thrombosis in patients with HIT, anticoagulation should be switched from heparin to an alternative anticoagulant; a direct thrombin inhibitor such as lepirudin (recombinant hirudin), bivalirudin, argatroban, fondaparinux, or danaparoid. Although not formally approved for this indication by the FDA, there are an increasing numbers of anecdotal reports of patients with HIT being successfully managed with the pentasaccharide, fondaparinux, in lieu of a direct thrombin inhibitor (DTI).12 The 2008 ACCP Guidelines give a stronger recommendation for the use of danaparoid, lepirudin, or argatroban in HIT than for fondaparinux.1 Because of their different modes of excretion and inactivation, patients with HIT and renal insufficiency are usually treated with argatroban, while those with hepatic impairment are usually treated with lepirudin. For patients in whom both renal and hepatic function are abnormal, treatment with argatroban, or bivalirudin (not otherwise approved for the treatment of HIT), at reduced doses is recommended.13,14
If thrombosis has not already been detected, duplex Doppler ultrasound of the lower extremities should be performed to rule out subclinical DVT. Platelet counts are followed daily. The DTI should be continued until the platelet count has recovered to at least 100 × 109/L, at which point treatment with a vitamin K antagonist (warfarin) may be initiated. The DTI should be continued until therapeutic anticoagulation with warfarin has been achieved (INR of 2.0-3.0). Warfarin is contraindicated as initial treatment of HIT because it may increase the risk of venous limb gangrene in patients with deep vein thrombosis through its rapid lowering of protein C levels.15 The length of treatment has not been defined but warfarin subsequently should be continued for at least 2-3 months due to a persistent risk of thrombosis even after the platelet count has recovered,16 whereas in patients in whom thrombosis has been documented (HITT), anticoagulation with warfarin should continue for at least 3-6 months.8 Heparin allergy should be documented in medical record (confirmed cases). Subsequent exposure to heparin should be avoided. If its use is regarded as necessary for a procedure, it should not be given until PF4-heparin antibodies are no longer detectable by ELISA (usually as of 100 days following an episode of HIT),17 and exposure should be limited to the shortest time period possible, but LMW heparin is not recommended.
Platelet transfusions are generally considered as being relatively contraindicated for the prevention of bleeding in patients with HIT, largely due to the possibility that they might precipitate thrombotic events. The 2008 ACCP Guidelines recommend that platelet transfusions can be considered in patients with HIT and overt bleeding or who are deemed to be at high bleeding risk, particularly if heparin has been stopped for at least several hours.1
Conclusion
HIT is not uncommon in hospital settings where heparin is used quite often. It is mostly a clinical diagnosis, and the main value of laboratory testing is in excluding the diagnosis. It can be easily missed or misdiagnosed particularly in complicated, critically ill patients with other multiple factors that can lead to thrombocytopenia, who may be receiving heparin for DVT prophylaxis. Platelet count should be monitored frequently during heparin administration and whenever thrombocytopenia occurs, it is imperative that pretest probability of HIT should be evaluated using the 4 T’s score. The diagnosis of HIT should be respected (hats off to HIT!) as it can lead to serious and potentially fatal complications. Heparin should be immediately substituted by an alternative anticoagulant, and laboratory confirmation should be planned along with Doppler study of the legs to rule out subclinical DVT. Heparin allergy should be clearly mentioned in the medical record of the patient.
Acknowledgements
The authors reported no conflict of interest and no funding was received for this work. |
|
| |
References
1. Warkentin TE, Greinacher A, Koster A, Lincoff AM; American College of Chest Physicians. Treatment and prevention of heparin-induced thrombocytopenia: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008 Jun;133(6)(Suppl):340S-380S.
2. Greinacher A. Antigen generation in heparin-associated thrombocytopenia: the nonimmunologic type and the immunologic type are closely linked in their pathogenesis. Semin Thromb Hemost 1995;21(1):106-116.
3. Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood 2005 Oct;106(8):2710-2715.
4. Smythe MA, Koerber JM, Mattson JC. The incidence of recognized heparin-induced thrombocytopenia in a large, tertiary care teaching hospital. Chest 2007 Jun;131(6):1644-1649.
5. Warkentin TE, Sheppard JA, Sigouin CS, Kohlmann T, Eichler P, Greinacher A. Gender imbalance and risk factor interactions in heparin-induced thrombocytopenia. Blood 2006 Nov;108(9):2937-2941.
6. Rauova L, Poncz M, McKenzie SE, Reilly MP, Arepally G, Weisel JW, et al. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005 Jan;105(1):131-138.
7. Newman PM, Chong BH. Heparin-induced thrombocytopenia: new evidence for the dynamic binding of purified anti-PF4-heparin antibodies to platelets and the resultant platelet activation. Blood 2000 Jul;96(1):182-187.
8. Alving BM. How I treat heparin-induced thrombocytopenia and thrombosis. Blood 2003 Jan;101(1):31-37.
9. Rice L, Attisha WK, Drexler A, Francis JL. Delayed-onset heparin-induced thrombocytopenia. Ann Intern Med 2002 Feb;136(3):210-215.
10. Lo GK, Juhl D, Warkentin TE, Sigouin CS, Eichler P, Greinacher A. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost 2006 Apr;4(4):759-765.
11. Selleng K, Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia in intensive care patients. Crit Care Med 2007 Apr;35(4):1165-1176.
12. Blackmer AB, Oertel MD, Valgus JM. Fondaparinux and the management of heparin-induced thrombocytopenia: the journey continues. Ann Pharmacother 2009 Oct;43(10):1636-1646.
13. Kiser TH, Fish DN. Evaluation of bivalirudin treatment for heparin-induced thrombocytopenia in critically ill patients with hepatic and/or renal dysfunction. Pharmacotherapy 2006 Apr;26(4):452-460.
14. Beiderlinden M, Treschan TA, Görlinger K, Peters J. Argatroban anticoagulation in critically ill patients. Ann Pharmacother 2007 May;41(5):749-754.
15. Srinivasan AF, Rice L, Bartholomew JR, Rangaswamy C, La Perna L, Thompson JE, et al. Warfarin-induced skin necrosis and venous limb gangrene in the setting of heparin-induced thrombocytopenia. Arch Intern Med 2004 Jan;164(1):66-70.
16. Warkentin TE, Kelton JG. A 14-year study of heparin-induced thrombocytopenia. Am J Med 1996 Nov;101(5):502-507.
17. Wanaka K, Matsuo T, Matsuo M, Kaneko C, Miyashita K, Asada R, et al. Re-exposure to heparin in uremic patients requiring hemodialysis with heparin-induced thrombocytopenia. J Thromb Haemost 2010 Mar;8(3):616-618. |
|