Within the spectrum of chronic renal disease, autosomal dominant polycystic kidney disease (ADPKD) accounts for one out of every 400–1000 live births, being a hereditary disorder with cystic and noncystic manifestations as well as extrarenal involvements.1 The latter has been widely reported comprising intestinal and meningeal diverticula, cardiac valve abnormalities, arterial dissection, and abdominal wall hernias.2
Pericardial involvement, although not as common as other extrarenal manifestations, might occur in patients with ADPKD. However, it is generally overlooked by physicians other than nephrologists and when discovered could result in excessive concern for both doctors and patients.3 Therefore, the knowledge of the insights of this extrarenal expression in such patients is crucial.
Case Report
We present the case of a 35-year-old female Moroccan patient with the diagnosis of ADPKD since childhood associated with arterial hypertension.
After being asymptomatic for over one year, the patient came into the Emergency Department with lower abdominal pain lasting for five days, vomiting with gastric content over the past 24 hours, and general fatigue. The patient denied fever, chills, arthralgia, cough, or myalgia. Upon interrogation and physical examination, the patient was initially diagnosed with salpingitis and was subsequently admitted in the hospital for confirmation of diagnosis and proper treatment. The results of her blood tests showed an elevation in urea 11.30 mmol/L (normal range: 2.76–8.07 mmol/L) and creatinine 92 mmol/L (normal range: 53–88 mmol/L).
Abdominal computed tomography (CT) scan with contrast was performed showing both kidneys normal in size with poor parenchymal definition associated to several cystic images with a thin wall, translucent content, without solid elements, intracystic hemorrhage, or sepsis sign [Figure 1] with clear and defined outline with a thickening at the left fallopian tube. A consult with a nephrologist established the need for a close follow-up.
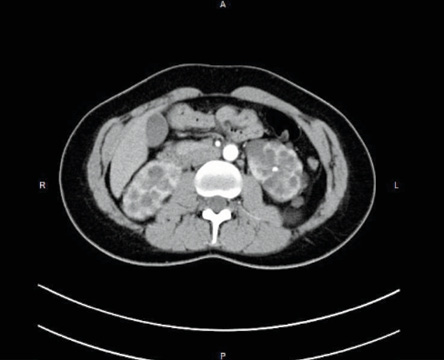
Figure 1: Abdominal computed tomography scan with contrast with several cystic images with a thin wall, translucent content, without solid elements or intracystic hemorrhage or sepsis sign.
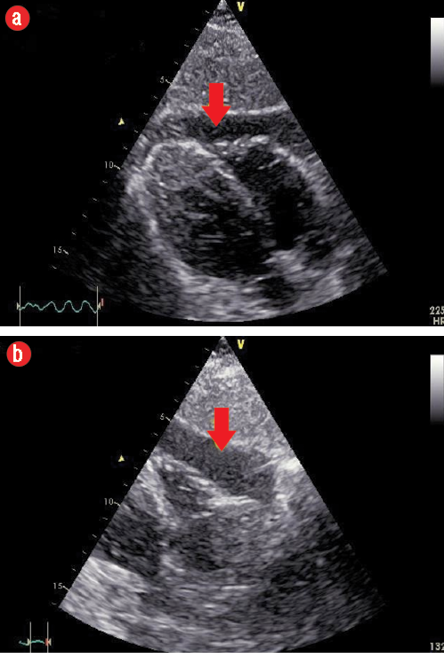
Figure 2: (a) Subcostal view showing mild to moderate anterior pericardial effusion (PE) extending towards the apex. (b) Subcostal view showing moderate PE at the level of the right ventricle free wall.
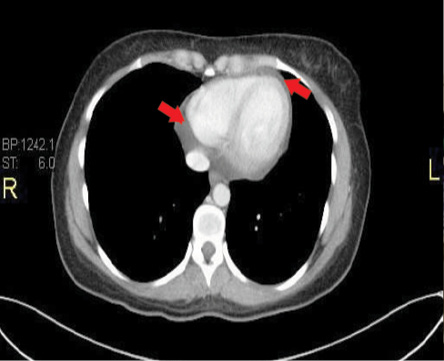
Figure 3: Representative transaxial thin computed tomography sections at low levels of the heart showing moderate pericardial effusion. Red arrows indicate the pericardial fluid.
After one day of hospitalization, the patient started complaining of palpitations with a heart rate of 115 beats per minute and dizziness when getting out of bed, which prevented the patient from standing. There was no hemodynamic instability during the three episodes of approximately five minutes duration over 24 hours.
One chest X-ray performed at that time showed no significant pathology. A transthoracic echocardiogram revealed the presence of moderate pericardial effusion (PE) more in the anterior aspect, extending to the apex with no signs of impending cardiac tamponade [Figure 2 a and b], the biventricular ejection fraction was normal.
In addition, a thorax CT scan with contrast confirmed the echocardiographic findings of a PE [Figure 3].
A greater set of standard tests to rule out collagen vascular disease, rheumatoid diseases, autoimmune disorders, and malignancies was ordered. These tests yielded no abnormalities.
The patient’s symptoms resolved spontaneously without any specific treatment. After five days, the patient was discharged with no symptoms and was advised for follow-up by both nephrology and cardiology specialties.
Two weeks after discharge, the patient had another echocardiogram which displayed no changes compared to the first one during hospitalization.
Discussion
Although ADPKD begins in-utero, it may take several decades for the symptoms and signs to be evident. As an autosomal dominant disorder, offspring possess a 50% risk of getting the mutated gene. However, only about half of these cases are diagnosed during life and might have an asymptomatic course.4
Two distinct genetic defects have been recognized in the pathogenesis of ADPKD: PKD1 which represents nearly 85% of the cases and PKD2 (10–15% of the cases).5
The polycystin complex (PKD1 and PKD2 encoding polycystin-1 and polycystin-2, respectively) is implicated in the regulation of the cell cycle and holds the key in the pathogenesis of ADPKD. Thus, a defective polycystin complex might lead to abnormal epithelial cell differentiation and the various phenotypic expressions of ADPKD.6
Among the cardiovascular manifestations of ADPKD, valvular abnormalities have been reported by cardiac ultrasonography in up to 30% of patients with ADPKD. The most frequent is mild mitral valve prolapse and aortic regurgitation, and less common mitral and/or tricuspid regurgitation.7
In addition, some reports suggest that ADPKD can also be associated with an increased incidence of coronary aneurysms.8
Asymptomatic PE appear to occur at increased frequency in patients with ADPKD. Spaia et al,9 reported a case of a 48-year-old woman, suffering from end-stage renal disease due to ADPKD who developed refractory pericarditis with moderate PE. In contrast, our patient had good renal function with well-tolerated PE and did not require any intervention.
Islam et al,10 performed consecutive CT scans in 60 patients with ADPKD, 100 patients without ADPKD with serum creatinine concentrations of 1.1 mg/dL, and 100 potential kidney donors who were retrospectively examined. Of those with ADPKD, 35% had PE compared with 9% of those without ADPKD. All PE were asymptomatic. Although frequently moderate to large, these effusions are generally well-tolerated and clinically inconsequential. In our case, likewise, the PE had no clinical consequences.
PE in the context of a patient with ADPKD is complex and is not entirely defined. Several theories have been proposed. The most accepted, so far, is linked to mutations in the PKD1 gene, which entails an abnormal production of matrix components, matrix-degrading enzymes, and inhibitors of metalloproteinases, and defects in connective tissue leading to an abnormal distensibility of the connective tissue (increased compliance and impaired recoil), possibly combined with a slightly increased extracellular volume facilitating the development of PE in patients with ADPKD.9,10
CT scan usage is increasingly expanding in medicine practice10,11 a well-established technique to detect the existence and severity of PE, although it is less sensitive than echocardiography to assess its hemodynamic impact. Echocardiography is still the best imaging technique to make a bedside assessment of any patient in which PE is suspected.12 Consequently, setting-up standard operating procedures and developing pathways for the clinicians to manage such patients will improve the quality of care and will undoubtedly reduce the patients’ exposure to radiation as well as the overall cost to the radiology department.13 Thus, this will help to minimize the growing demands on the radiology department.
In our patient, the PE was graded as mild to moderate with no compromise of the hemodynamics but raised substantial concern on the doctors who looked after the patient. Thus, the patient was subjected to different tests to find out the cause of the pericardial affectation. Even the likelihood of a pericardial biopsy was considered, however, this choice was dismissed after the consultation with a nephrologist.
Conclusion
PE is an extrarenal manifestation in the setting of a patient with ADPKD present in up to 35% of such patients. Awareness of this co-existence, in the absence of hemodynamic compromise, should prevent excessive diagnostic tests by the physicians.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007 Apr;369(9569):1287-1301.
- 2. Catapano F, Pancaldi S, Pace Napoleone C, De Sanctis LB, Gargiulo G, Emiliani G, et al. An unusual cardiac manifestation in autosomal dominant polycystic kidney disease. Case Rep Nephrol 2012;2012:978170.
- 3. Schrier RW. Optimal care of autosomal dominant polycystic kidney disease patients. Nephrology (Carlton). 2006 Apr;11(2):124-30.
- 4. Wilson PD. Polycystic kidney disease. N Engl J Med 2004 Jan;350(2):151-164.
- 5. Davies F, Coles GA, Harper PS, Williams AJ, Evans C, Cochlin D. Polycystic kidney disease re-evaluated: a population-based study. Q J Med 1991 Jun;79(290):477-485.
- 6. Chen WC, Tzeng YS, Li H. Gene expression in early and progression phases of autosomal dominant polycystic kidney disease. BMC Res Notes 2008 Dec;1:131.
- 7. Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988 Oct;319(14):907-912.
- 8. Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998 May;9(5):837-841.
- 9. Spaia S, Patsalas S, Agelou A, Theodoroglou C, Askepidis N, Pazaroglou M, et al. Managing refractory uraemic pericarditis with colchicine. Nephrol Dial Transplant 2004 Sep;19(9):2422-2423.
- 10. Islam SM, Abru AF, Al Obaidani S, Shabibi SA, Al Farsi S. Trends in CT request and related outcomes in a Pediatric Emergency Department. Oman Med J 2016 Sep;31(5):365-369.
- 11. Bagga R, Rai R, Kalra J, Saha PK, Singh T. An unusual cause of postabortal fever requiring prompt surgical intervention: a pyomyoma and its imaging features. Oman Med J 2017 Jan;32(1):73-76.
- 12. Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2007 Nov;2(6):1223-1227.
- 13. Al-Nabhani K, Kakaria A, Syed R. Computed tomography in management of patients with non-localizing headache. Oman Med J 2014 Jan;29(1):28-31.