Langerhans cell histiocytosis (LCH) is a rare proliferative disorder with unknown etiologies.1,2 Immunoregulatory defects leading to abnormal maturation and migration of the Langerhans cells to the central nervous system, lungs, skin, bone, bone marrow, lymph nodes, thymus, liver, and spleen were postulated to be the causative factors for LCH.3 The incidence of LCH is about five to six cases per one million children.4
Males are more frequently affected than females, and the average age of presentation varies from a few months to 15 years.3 It can present as solitary or multifocal lesions5 and may involve multiple systems. Head and neck involvement is found in almost 60% of cases and is the most common site for LCH.3 Pertaining to otorhinolaryngology entity, the disease incidences in various sites were reported to be 44–48% in the skull vault and orbit, 21–26% in the cervical lymph nodes, 19–25% in the temporal bone, and 7–10% in the upper and lower jaw.3
As the presentation of otologic histiocytosis may mimic those of acute and chronic infectious diseases,1 diagnosis can be difficult. This, in turn, may lead to a delay in treatment and thus affect the overall prognosis. LCH is diagnosed based on typical histopathology findings,6 and only tissue biopsy can lead to a definitive diagnosis.1 Computed tomography (CT) can be useful in defining the lesion and extent of temporal bone involvement.6
Case report
A one-year-old girl presented to us with intermittent bilateral otorrhoea for four months. Despite multiple courses of topical and systemic antibiotics, her symptoms persisted. She returned with a left postauricular swelling and pyrexia for one week. There was no history of chronic nasal discharge or upper respiratory tract infection during her initial presentation.
Clinical examination revealed a firm, warm, tender, and erythematous swelling over the left postauricular region [Figure 1]. The swelling measured 2 × 2 cm and displaced her left pinna anteroinferiorly. Bilateral otoscopic examination displayed granulation tissue mixed with a copious amount of mucopurulent discharge, which was obscuring the tympanic membrane view. She also had a small ulcerative lesion on the hard palate. Multiple firm, non-tender subcentimeter cervical lymph nodes were palpable bilaterally.
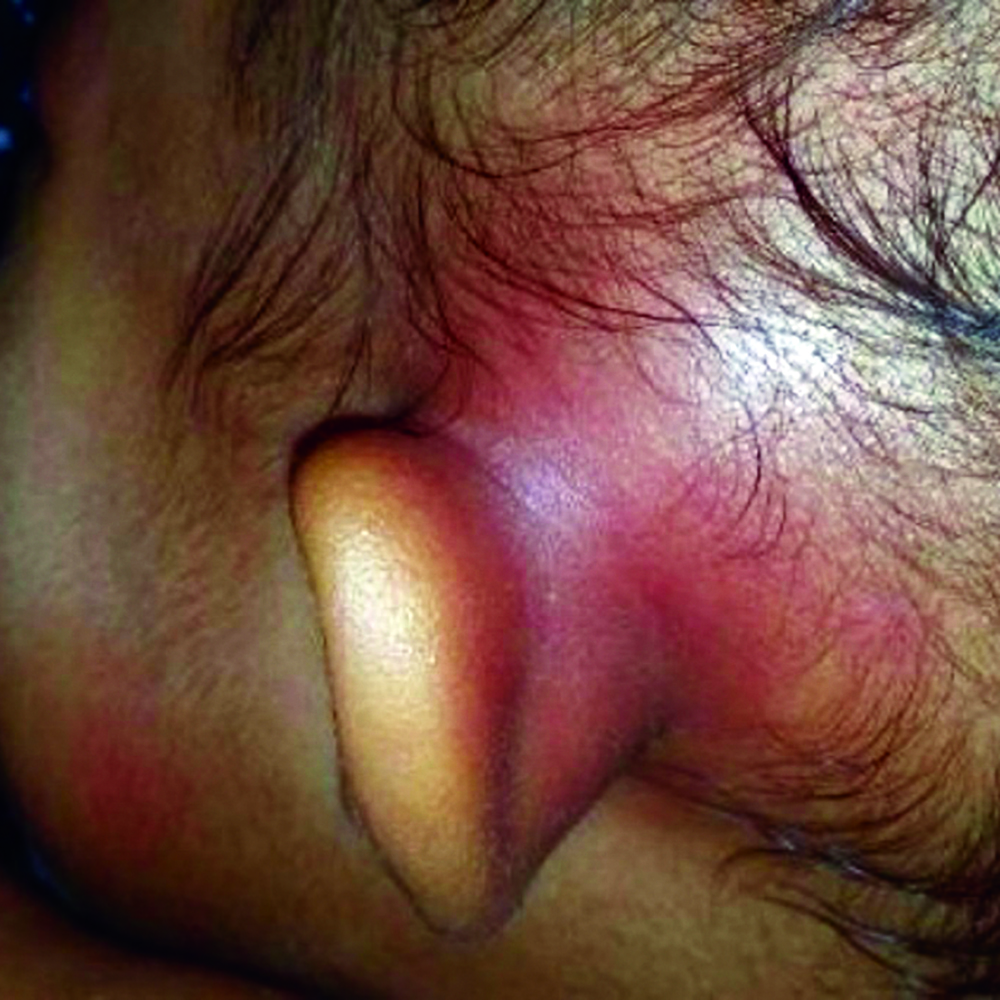
Figure 1: Swelling over the left postauricular region suggestive of acute mastoiditis or mastoid abscess.
CT imaging was suggestive of bilateral mastoiditis with collections over the left mastoid region and demonstrated erosion of the left tegmen tympani with no intracranial extension [Figure 2]. However, the isodensity of the lesion could also suggest the possibility of a soft tissue tumor. She was admitted for systemic antibiotic therapy and subsequently underwent examination under anesthesia. Intraoperative findings revealed a soft tissue mass occupying the mastoid cavity with serous fluid collection. Tissue biopsies taken from the mass, external ear canal, and the hard palate eventually confirmed the diagnosis of LCH with the detection of CD1a and S-100 protein [Figure 3]. Treatment modalities, including chemotherapy, were discussed. Unfortunately, her parents refused further intervention and opted for conservative management. They did not present for follow-up subsequently.
Differential diagnosis
A differential diagnosis of mastoiditis and cholesteatoma with mastoid abscess was made.
The patient, unfortunately, succumbed to the illness a few weeks after the diagnosis. The cause of death was not ascertained as the family members refused a post-mortem.
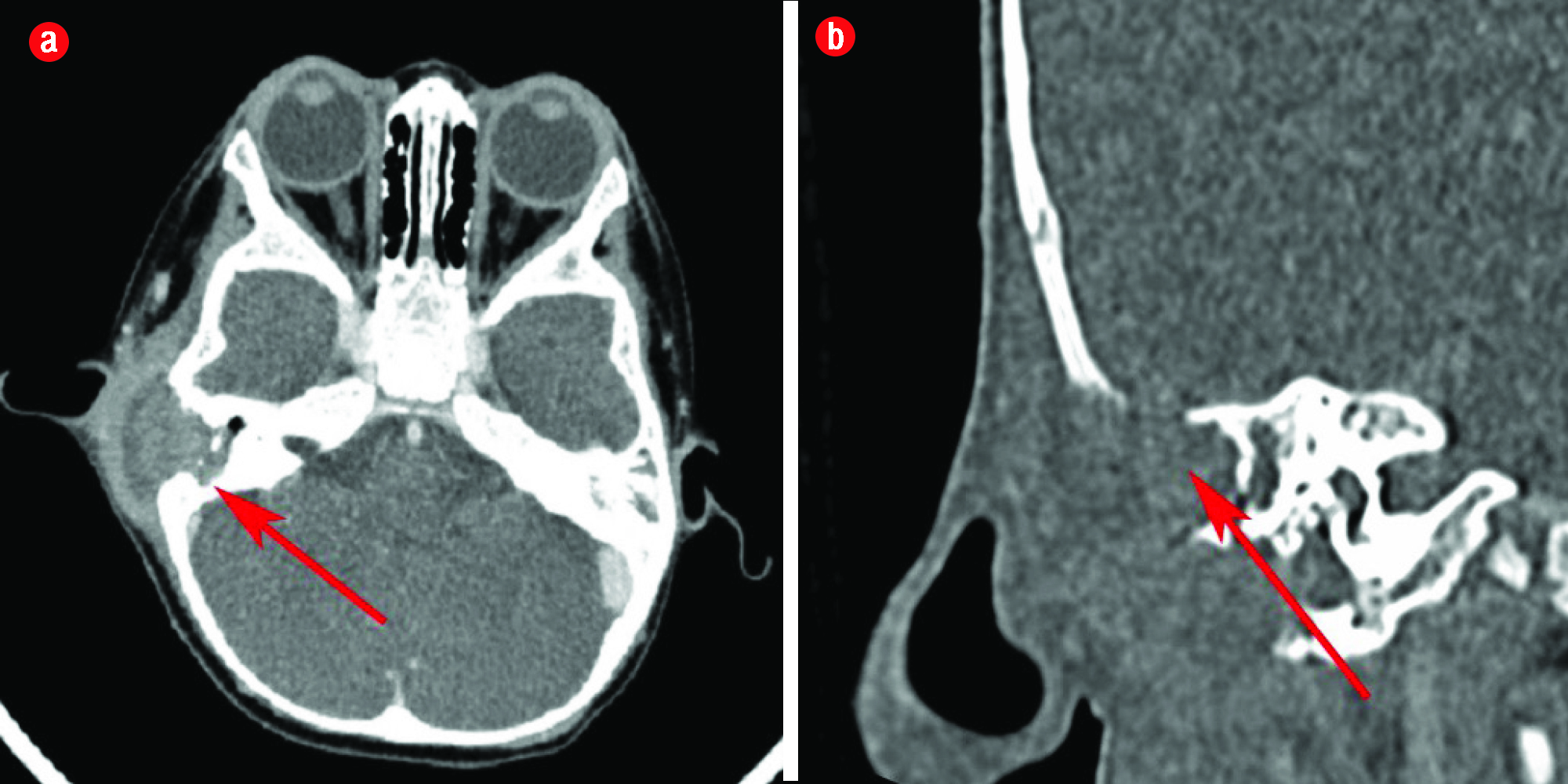
Figure 2: (a) Axial and (b) coronal view of the computed tomography scan done on the patient showing an abscess formation on her left temporal region with erosion.
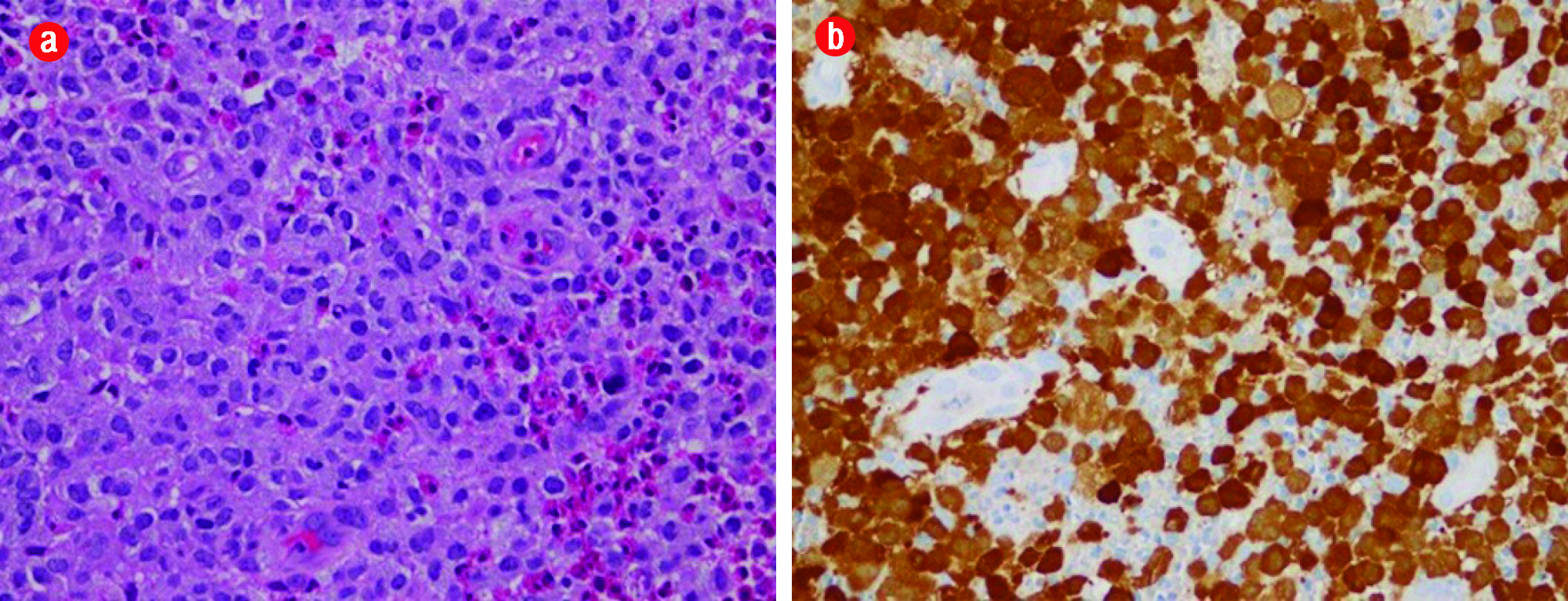
Figure 3: (a) Microscopic view of Langerhans cells (hematoxylin and eosin stain) exhibiting large, pale, and folded vesicular nuclei and abundant eosinophilic cytoplasm. (b) Langerhans cells positive for S-100 (brown staining of the nucleus and cytoplasm) and CD1a (brown staining of cytoplasmic membrane), magnification = 40 ×.
Discussion
LCH has been well known to mimic a variety of common diseases and misdiagnosis is not uncommon.7 There is a wide clinical spectrum, ranging from isolated lesions to multisystem involvement with the bone tissue most commonly affected.5 Moreover, studies have shown that bone marrow involvement is more likely to occur in patients with multisystem involvement resulting in cytopenia.5 Our patient had bilateral temporal bone involvement and hard palate involvement. Oral involvement of LCH usually presents as swelling or ulceration, with or without the involvement of the underlying bone.7
The presence of atypical otitis media, otorrhoea, aural polyps, granulations tissues, or a postauricular mass should alert to the possibility of LCH.6 Our patient presented initially with intermittent bilateral otorrhoea for four months and therefore was treated as a case of otitis media. However, she was resistant to treatment. She subsequently developed a left postauricular swelling with spiking fever that was suggestive of mastoid cellulitis. CT imaging revealed bilateral temporal bone involvement with sparing of the bony labyrinth, which is consistent with the diagnosis of LCH.4 Another important feature that may be strongly suggestive of LCH, is the presence of lytic or ‘punched out’ lesions of the temporal bone,4 which was not seen in our case. Magnetic resonance imaging (MRI) should be employed following CT imaging for further characterization of the lesion and to evaluate intracranial involvement.4 MRI was not done in this case due to the request of the family members for conservative management. The diagnosis of LCH was made based on histopathology results. It was supported by the detection of CD1a and S-100 protein. These markers are specific and helpful in differentiating LCH from granulomatous osteomyelitis or malignant lymphoma, which have similar histological properties.1 This implies that clinical and imaging tools are not adequate for accurate diagnosis and histopathological studies play an important role in the diagnosis.
Various treatment modalities have been used for LCH including close monitoring, surgical intervention, local steroids injections, high-dose systemic corticosteroids, low-dose radiotherapy, chemotherapy, bone marrow transplantation, and antibody therapy.7 Nonetheless, the combination of steroids and vinblastine has been accepted internationally as the standard treatment for LCH, even in patients with multisystem involvement.8 Generally, the overall prognosis is good but patients with multisystem involvement and active disease, such as seen in this case, tend to have a poorer outcome.6 In fact, the Histiocyte Society has categorized LCH patients into low- and high-risk groups, based on the outcomes related to the extent and the site of the LCH lesions.8 Patients with lungs, liver, spleen, and bone marrow involvement are considered to have higher risks and poorer outcomes. Furthermore, the presented case had features of tegmen tympani erosion, which is suggestive of a central nervous system risk lesion. This predisposes the child to severe, irreversible neurodegenerative disease.9
Obtaining a diagnosis of LCH can be challenging as its presentation may mimic a large number of diseases. In its early stages, a child may present with non-specific symptoms. Furthermore, tumors of the middle ear and temporal bone are uncommon and may be missed. A strong index of suspicion is important especially in children who present atypically for common diseases and are resistant to preliminary treatment. Timely diagnosis may ensure a better outcome as the prognosis of LCH is good when detected early.
Conclusion
The diagnosis of LCH should be considered in children as a differential when they present with chronic otorrhoea with the presence of a mass or granulation tissue, which does not respond well to conventional medical treatment. Full systemic examination is warranted in children who do not respond to medical treatment to exclude multisystemic involvement of the disease and to detect if the child is immunocompromised. As CT imaging poses children to radiation exposure, MRI may be considered as an alternative. A deep tissue biopsy is indicated in suspicious lesions or cases resistant to treatment.
Disclosure
The authors declared no conflicts of interest.
Acknowledgements
The authors would like to acknowledge Dr. Johnson Ng Wei Siang and the operating team of the Department of Otorhinolaryngology of Hospital Tengku Ampuan Rahimah, Klang, Malaysia for their contributions to the realization of this manuscript.
references
- 1. Kleinjung T, Woenckhaus M, Bachthaler M, Wolff JE, Wolf SR. Langerhans’ cell histiocytosis with bilateral temporal bone involvement. Am J Otolaryngol 2003 Jul-Aug;24(4):265-270.
- 2. Rodríguez Rivera V, Lesmas Navarro MJ, de Paula Vernetta C, Donderis Sala J, Morera Faet H. [Early start eosinophilic granuloma of the temporal bone]. Acta Otorrinolaringol Esp 2010 Jul-Aug;61(4):315-317.
- 3. Martini A, Aimoni C, Trevisani M, Marangoni P. Langerhans’ cell histiocytosis: report of a case with temporal localization. Int J Pediatr Otorhinolaryngol 2000 Sep;55(1):51-56.
- 4. Mira AC, Jane M, Carrie MC, Laurence JE, Amulya ANR.Bilateral temporal bone Langerhan cells histiocytosis: Radiologic pearls. Open Neuroimaging J 2013;7:53-57
- 5. Galluzzo ML, Braier J, Rosenzweig SD, Garcia de Dávila MT, Rosso D. Bone marrow findings at diagnosis in patients with multisystem langerhans cell histiocytosis. Pediatr Dev Pathol 2010 Mar-Apr;13(2):101-106.
- 6. Semih M, Erkan K, Cengiz D, Kanlikama M, Sirikci A, Bakir K, et al. A rare disorder mimics otitis media in children:Langerhans’ cells histiocytosis. Int J Pediatr Otorhinolaryngology Extra 2007;70(10):68-71.
- 7. Kilic E, Er N, Mavili E, Alkan A, Gunhan O. Oral mucosal involvement in Langerhans’ cell histiocytosis: long-term follow-up of a rare case. Aust Dent J 2011 Dec;56(4):433-436.
- 8. Filipovich A, McClain K, Grom A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant 2010 Jan;16(1)(Suppl):S82-S89.
- 9. Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 2013 Feb;60(2):175-184.