Carnitine-acylcarnitine translocase deficiency (CACTD) is a rare and life-threatening autosomal recessive disorder of mitochondrial fatty acid β-oxidation (FAO) caused by a variation of the SLC25A20 gene on chromosome 3p21.31.1 The significantly increased acylcarnitine profiles are detected in dry blood spots by tandem mass spectrometry.2 At least 42 different pathogenic or possibly pathogenic variants of SLC25A20 have been identified to date that cause CACTD. In Asia, the c.199-10T>G splice site variation is the most frequently reported. This metabolic disease is rare; it leads to life-threatening conditions, with an estimated incidence of 1/60 000 in Hong Kong.3
Ryder et al,4 reported approximately 87 cases related to this metabolic disorder. Most patients present in the first two days of life, with hypoketotic hypoglycemia, hyperammonemia, cardiomyopathy or arrhythmia, hepatomegaly, and elevated liver enzymes.4,5 Despite widely differing clinical manifestations of CACTD, two distinct clinical subtypes exist: a neonatal-onset severe form and an infancy-onset milder form.6 Autopsy and histopathological examination often reveal extensive vascular degeneration in the heart and liver.3,7 Gene mutation detection remains the gold standard for diagnosing CACTD.7
In the context of genetic advances, preimplantation genetic testing (PGT) has become a well-established alternative to invasive prenatal diagnosis, especially for monogenic disorders. PGT involves biopsying one or a few cells from in vitro fertilization embryos, testing the samples for genetic aberrations, and selectively transferring embryos without the identified genetic condition. While PGT is a suitable solution for couples at high risk of transmitting known genetic conditions, its application is currently limited in low-resource settings.8
This case report contributes valuable insights into a rare disease associated with the SLC25A20 gene mutation and highlights the significance of genetic screening and embryonic selection in low- and middle-income countries.
Case report
A 26-year-old pregnant woman (G5P2) was admitted to our hospital for a term pregnancy with an unremarkable medical record. Her obstetric history included one stillbirth at eight weeks of gestation and two full-term deliveries by vaginal birth and cesarean section, with neonates weighing 3000 and 3100 g, respectively. Unfortunately, both newborns died suddenly within two days of birth, and the etiology remained unexplained. The clinical presentations of the neonates included rapid deterioration, poor response, low muscle strength and tone, cyanosis, cardiopulmonary collapse, and eventual cardiac arrest despite resuscitation efforts. Both parents denied consanguineous marriage, and an investigation into the family history revealed no similar sudden deaths. Parental carrier testing showed a heterozygous SLC25A20 c.199-10T>G mutation in both mother and father. Additionally, the father had gene mutations in MBL2 and IDS, and the mother had a variation in the BCKDHB gene. Despite these genetic findings, both parents were asymptomatic. Two years prior, the patient underwent a medical termination of pregnancy at 20 weeks of gestational age due to homozygosity for the c.199-10T>G variant of the SLC25A20 gene. Confirmatory diagnosis was obtained through amniocentesis, and this pregnancy was conceived using artificial reproductive technology with PGT. Upon hospitalization, the patient’s vital signs were stable, and she showed no signs of labor during examination. Sonographic findings revealed a vital fetus corresponding to 39 weeks and two days of gestational age. Fetal heart monitoring was normal, and laboratory tests fell within the normal range. Initially scheduled for induction of labor, the patient underwent cesarean section due to failed induction of labor and a history of adverse obstetric events. A male neonate was promptly assessed at birth, with Apgar scores of eight at one minute and nine at five minutes. The newborn, weighing 2900 g, appeared normal, and initial serum analyses showed a slight electrolyte imbalance (Na+: 131 mmol/l, K+: 6.02 mmol/l, Cl-: 85.5 mmol/l, Ca2+: 2.08 mmol/l, NH3-: 109 umol/l), which were rapidly corrected. The neonate was monitored for seven days in the newborn care unit without complications. Both the mother and baby were discharged, with the baby being strictly monitored postnatally. Blood and urine samples were collected for tandem mass spectrometry and gas chromatography-mass spectrometry analyses.
Gas chromatography-mass spectrometry analysis of urine (QP2020 system, Shimadzu, Japan) revealed slightly elevated values of glycolic acid and palmitic acid at 5.7% (normal limit: < 4.0%) and 43.3% (normal limit: < 23.3%), respectively [Figure 1]. However, no metabolic diseases were detected in the blood samples [Table 1]. Subsequent blood tests for acylcarnitine, amino acids, hemoglobin abnormalities, and other rare metabolic pathologies showed normal results.
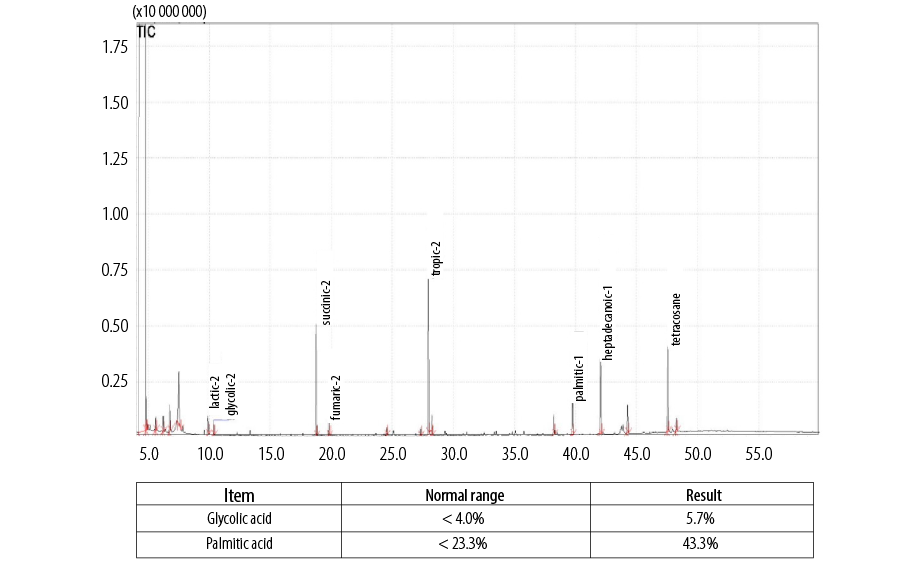 Figure 1: Gas chromatography-mass spectrometry analysis of urine specimen, showing slightly higher values of glycolic acid and palmitic acid than the normal limit.
Figure 1: Gas chromatography-mass spectrometry analysis of urine specimen, showing slightly higher values of glycolic acid and palmitic acid than the normal limit.
Table 1: Results of blood specimen tests indicating the absence of metabolic diseases related to acylcarnitine, amino acids, hemoglobin abnormalities, and other rare metabolic pathologies.
|
Glucose-6-phosphate dehydrogenase
|
118.74
|
> 41
|
µM NADH
|
Normal
|
|
Thyroid stimulating hormone
|
0
|
< 30
|
µIU/mL
|
Normal
|
|
17-hydroxyprogesterone
|
3.6
|
< 30
|
ng/mL
|
Normal
|
|
Phenylalanine
|
1.41
|
< 3.9
|
mg/dL
|
Normal
|
|
Total galactose
|
1.42
|
< 13
|
mg/dL
|
Normal
|
|
Thyroxine
|
13.5
|
4–22.6
|
µg/dL
|
Normal
|
|
Galactose-l-phosphate uridyltransferase
|
4.15
|
> 2.0
|
U/g Hb
|
Normal
|
|
Biotinidase
|
91.14
|
> 36
|
MRU
|
Normal
|
|
Pathologies relating to hemoglobin
(Thalassemia and variation of hemoglobin disease (≥ 5 diseases)
|
Low risk
|
|
|
|
|
Amino acid
|
|
|
|
|
|
Alanine
|
120.6
|
0–550
|
µmol/L
|
Normal
|
|
Arginine
|
12.2
|
0–36
|
µmol/L
|
Normal
|
|
Aspartic
|
83.25
|
0–810
|
µmol/L
|
Normal
|
|
Citruline
|
11.53
|
3.5–40
|
µmol/L
|
Normal
|
|
Glutamic acid
|
322.86
|
0–1000
|
µmol/L
|
Normal
|
|
Glycine
|
136.49
|
0–470
|
µmol/L
|
Normal
|
|
Leucine
|
175.39
|
30–280
|
µmol/L
|
Normal
|
|
Methionine
|
19.13
|
3.5–30
|
µmol/L
|
Normal
|
|
Ornithine
|
109.93
|
20–245
|
µmol/L
|
Normal
|
|
Phenylalanine
|
46.78
|
0–125
|
µmol/L
|
Normal
|
|
Proline
|
218.01
|
60–410
|
pmol/L
|
Normal
|
|
Tyrosine
|
126.67
|
0–210
|
µmol/L
|
Normal
|
|
Valine
|
109.83
|
20–200
|
µmol/L
|
Normal
|
|
Free carnitine and acylcarnitine
|
|
|
|
|
|
Free carnitine (CO)
|
19.83
|
8–110
|
µmol/L
|
Normal
|
|
Acetylcarnitine (C2)
|
11.95
|
2.3–45
|
µmol/L
|
Normal
|
|
Propionylcarnitine (C3)
|
0.86
|
0.3–6
|
µmol/L
|
Normal
|
|
Butyrylcarnitine (C4)
|
0.12
|
0–0.61
|
µmol/L
|
Normal
|
|
C3DC + C40H
|
0.02
|
0–0.14
|
µmol/L
|
Normal
|
|
C4DC + C50H
|
0.06
|
0–0.5
|
µmol/L
|
Normal
|
|
Isovalerycarnitine (C5)
|
0.07
|
0–0.5
|
µmol/L
|
Normal
|
|
Tyglycarnitine (C5: I)
|
|
0–0.06
|
µmol/L
|
Normal
|
|
C5DC + C60H
|
0.12
|
0–0.45
|
µmol/L
|
Normal
|
|
Hexanoylcarnitine (C6)
|
0.03
|
0–0.14
|
µmol/L
|
Normal
|
|
Octanoylcarnitine (C8)
|
0.03
|
0–0.2
|
µmol/L
|
Normal
|
|
Octenoylcarnitine (C8:l)
|
0.03
|
0–0.25
|
µmol/L
|
Normal
|
|
Decanoylcarnitine (CIO)
|
0.03
|
0–0.17
|
µmol/L
|
Normal
|
|
Decenoylcarnitine (Cl O: l)
|
0.02
|
0–0.16
|
µmol/L
|
Normal
|
|
Decadienoylcarnitine (CI 0:2)
|
0.02
|
0–0.09
|
µmol/L
|
Normal
|
|
Dodecanoylcarnitine (Cl 2)
|
0.03
|
0–0.25
|
µmol/L
|
Normal
|
|
Dodecenoylcarnitine (Cl 2:1)
|
0.01
|
0–0.26
|
µmol/L
|
Normal
|
|
Tetradecanoylcarnitine (C14)
|
0.11
|
0–0.5
|
µmol/L
|
Normal
|
|
Tetradecenoylcarnitine (C14:l)
|
0.04
|
0.01–0.3
|
µmol/L
|
Normal
|
|
Tetradecandienoylcarnitine (C14:2)
|
0.01
|
0–0.05
|
µmol/L
|
Normal
|
|
3-hydroxy-tetradecanoyIcarnitine (C140H)
|
|
0–0.03
|
µmol/L
|
Normal
|
|
Hexadecanoylcarnitine (Cl 6)
|
1.46
|
0.3–6
|
µmol/L
|
Normal
|
|
Hexadecenoylcarnitine (C16: I)
|
0.08
|
0.01–0.4
|
µmol/L
|
Normal
|
|
3-hydroxy-hexadecenoyIcarnitine (CI OH)
|
0.03
|
0–0.08
|
µmol/L
|
Normal
|
|
Hexadecadienoylcarnitine (Cl 6:2)
|
0
|
0–0.03
|
µmol/L
|
Normal
|
|
3-hydroxy-hexadecanoyIcarnitine (CI 60H)
|
0.01
|
0–0.05
|
µmol/L
|
Normal
|
|
Octadecanoylcarnitine (C18)
|
0.58
|
0.18–1.9
|
µmol/L
|
Normal
|
|
Octadecenoylcarnitine (C18:I)
|
0.77
|
0.38–2.5
|
µmol/L
|
Normal
|
|
3-hydroxy-octadecenoyIcarnitine (Cl 8:101-1)
|
0.01
|
0–0.05
|
µmol/L
|
Normal
|
|
Octadecadienoylcarnitine (C18:2)
|
0.13
|
0.03–0.8
|
µmol/L
|
Normal
|
|
3-hydroxy-Octadecadienoylcarnitine (CI 8:201-1)
|
0.01
|
0–0.04
|
µmol/L
|
Normal
|
At the time of this report, the baby remained in good condition without notable complications, and the family expressed gratitude for the positive outcomes. Ethical approval was waived for publication, all patient details were de-identified, and written informed consent was obtained from the parents. This case report adheres to the CARE guidelines.9
Discussion
In this case, the recurrence of two unexplained neonatal sudden deaths led to a comprehensive genetic analysis, revealing an autosomal recessive disorder associated with the SLC25A20 gene, confirming the diagnosis of CACTD. The severe phenotype observed in the neonatal deaths aligns with the typical clinical manifestations of CACTD, characterized by hypoketotic hypoglycemia, hyperammonemia, liver function damage, and elevated creatine kinase.5 Pathological changes include heart failure, arrhythmia, respiratory collapse, and cardiac arrest relating to the accumulation of long-chain fatty acids in multiorgan due to mitochondrial fatty acid β-oxidation disorders, which are immediately the direct cause of death. At the same time, gene mutation is the underlying cause of death [Table 2]. Neonatal death has mostly been noted after delivery or in the first week of life.5,10,11 Rare reports, such as Chen et al’s,3 describe late-onset cases emerging 61 days after birth.
Table 2: Cases of carnitine-acylcarnitine translocase deficiency with SLC25A20 c.199-10T>G variation in the last five years.
|
Yan et al,11 2017, China
|
-G3P3
-First infant (boy) died at 2 days with sudden cardiac death
|
25 minutes after birth
|
- Severe metabolic crisis
-Clinical conditions deteriorate rapidly
- Both died of cardiorespiratory collapse in the first week of life
|
Homozygous
|
Heterozygous status for the c.199-10T>G mutation
|
- High glucose and arginine infusion
- Respiratory and circulatory support
|
- Male
- Spontaneous VB
|
78 hrs
|
|
-
|
At 52 hours after birth
|
- Poor response and cyanosis
-Died of congestive heart failure
|
A compound heterozygous for two mutations: a novel c.1A>G mutation and a previously described c.199-10T>G mutation
|
The c.199-10T>G was derived from the maternal allele while the c.1A>G from the paternal allele
|
- Antishock therapy
- Arginine infusion
- Mechanical ventilation
|
- Female
- CS
- Apgar score of 10 pts at one minute
|
6 days
|
|
Chen et al,3 2020, China
|
-G4P3
-the second infant (boy) died on the day of birth of an unknown cause.
-The third child (girl)
is in good health
|
At 61 days of birth
|
- Severe metabolic crisis,
-Clinical condition rapidly deteriorated
- Respiratory insufficiency and cardiac arrest
|
Homozygous
|
Both parents and older sister were heterozygous
|
Several resuscitation attempts failed
|
- 36 wks
- Female
- CS
- 2200 g
|
61 days
|
|
Li et al,6 2021, China
|
-G2P1
-The first infant died at two days old from asphyxia, arrhythmia, and cardiac arrest
|
After 28 hours of birth
|
- Sleepy, no need of breastfeeding
- Ventricular tachycardia, bradycardia, and complete right bundle branch block between the ages of 47 and 51 hours
|
Homozygous
|
Both parents were heterozygous carriers of the variation
|
Resuscitation
|
- Full-term
- CS
- Apgar score 10 pts at 5 mins
|
3 days
|
|
Li et al,7 2022, China
|
Primipara
|
At two days of birth
|
- Hypnesthesia, convulsions, hypothermia and bradypnea
-Severe metabolic crisis
- Deteriorated rapidly
|
The parents were carriers of the gene mutation
|
A compound heterozygote with c.199–10 T>G and a novel c.1A> T mutation in the SLC25A20 gene
|
Resuscitation
|
- Full-term
- Spontaneous VB
- Male
- Normal birth weight
- Apgar score of 10 pts at 1 min
|
3 days
|
|
Zhang et al,5 2023, China
|
G1P2
|
a poor response, hypoglycemia, hypotonia, arrhythmias and sudden cardiorespiratory arrest on day 1.5
|
Hypoglycemia, arrhythmia, and sudden death
|
Two heterozygous variants of the SLC25A20 gene in the two infants: paternal variant M1:c.706_707insT: p.R236L fs*12 and maternal variant M2: c.689C>G:p.P230R
|
-Heterozygous status
- M1 variant was paternal
- M2 variant was maternal.
|
Cardiopulmonary resuscitation for 1 hr
|
- 37 wks 6 days
- CS
- Male-female twin
- 3490-3490 g
|
1.5–3.5 days
|
|
On 17th of life
|
- Sleeping until the 21st hour of life without waking to feed
- No spontaneous eye opening and had fair cry
- Generalized cyanosis and subsequently went into cardiac arrest
|
Missed
|
Both parents were identified to be heterozygous carriers of a pathogenic variant c.199-10T>G in the SLC25A20 gene.
|
Resuscitation
|
-37 wks
-CS due to non-reassuring fetal status
- Male
- 2400 g
- Good cry
|
33rd hr of life
|
CS: cesarean section; P: parity; G: gravida; VB: vaginal birth; wks: weeks; hr: hours; NICU: neonatal intensive care unit; IV: intravenous.
In assisted reproductive techniques, preimplantation genetic diagnosis offers couples with heritable genetic disorders a means to avoid the birth of a diseased offspring.13 In this case, in vitro fertilization with selective embryo transfer successfully avoided the birth of homozygous genetic carrier fetuses, resulting in a newborn carrying a heterozygous SLC25A20 gene without severe symptoms after birth. Carmona et al,12 also agreed that the reproductive choices through preimplantation genetic testing or through early confirmatory testing for CACTD in the neonates and anticipatory management could help improve severe neonatal outcomes.
The multidisciplinary approach, incorporating molecular diagnosis, prenatal screening, and neonatal care, represents the current standard for managing CACTD. Timely intervention is crucial in limiting neonatal morbidity and mortality associated with this life-threatening disorder.4
Conclusion
Obstetricians should maintain a high index of suspicion of CACTD, particularly in cases of recurrent unexplained neonatal deaths. Parental carrier testing is essential for prenatal management, and the use of selective embryos provides a viable option for heterozygous SLC25A20 gene-carried parents in this highly lethal disorder.
Disclosure
The authors declare no conflicts of interest. Ngoc Bich Trinh and Phuc Nhon Nguyen contributed equally to this paper and should be considered as co-first authors.
Acknowledgments
The authors would like to thank the patient and her family for sharing the clinical data for publication. The authors are also thankful to all the colleagues who took care of the patient and her newborn baby.
references
- 1. Palmieri F, Scarcia P, Monné M. Diseases caused by mutations in mitochondrial carrier genes SLC25: a review. Biomolecules 2020 Apr;10(4):655.
- 2. Habib A, Azize NA, Rahman SA, Yakob Y, Suberamaniam V, Nazri MI, et al. Novel mutations associated with carnitine-acylcarnitine translocase and carnitine palmitoyl transferase 2 deficiencies in Malaysia. Clin Biochem 2021 Dec;98:48-53.
- 3. Chen M, Cai Y, Li S, Xiong H, Liu M, Ma F, et al. Late-onset carnitine-acylcarnitine translocase deficiency with SLC25A20 c.199-10T>G variation: case report and pathologic analysis of liver biopsy. Front Pediatr 2020 Oct;8:585646.
- 4. Ryder B, Inbar-Feigenberg M, Glamuzina E, Halligan R, Vara R, Elliot A, et al. New insights into carnitine-acylcarnitine translocase deficiency from 23 cases: management challenges and potential therapeutic approaches. J Inherit Metab Dis 2021 Jul;44(4):903-915.
- 5. Zhang L, Hu Y, Xie M, Zhang Y, Cen K, Chen L, et al. Carnitine-acylcarnitine translocase deficiency caused by SLC25A20 gene heterozygous variants in twins: a case report. J Int Med Res 2023 Apr;51(4):3000605231163811.
- 6. Li X, Shen J. One potential hotspot SLC25A20 gene variants in Chinese patients with carnitine-acylcarnitine translocase deficiency. Front Pediatr 2022 Nov;10:1029004.
- 7. Li X, Zhao F, Zhao Z, Zhao X, Meng H, Zhang D, et al. Neonatal sudden death caused by a novel heterozygous mutation in SLC25A20 gene: a case report and brief literature review. Leg Med (Tokyo) 2022 Feb;54:101990.
- 8. De Rycke M, Berckmoes V. Preimplantation genetic testing for monogenic disorders. Genes (Basel) 2020 Jul;11(8):871.
- 9. Gagnier JJ, Kienle G, Altman DG, Moher D, Sox H, Riley D; CARE Group*. The care guidelines: consensus-based clinical case reporting guideline development. Glob Adv Health Med 2013 Sep;2(5):38-43.
- 10. Tang C, Liu S, Wu M, Lin S, Lin Y, Su L, et al. Clinical and molecular characteristics of carnitine-acylcarnitine translocase deficiency: experience with six patients in Guangdong China. Clin Chim Acta 2019 Aug;495:476-480.
- 11. Yan HM, Hu H, Ahmed A, Feng BB, Liu J, Jia ZJ, et al. Carnitine-acylcarnitine translocase deficiency with c.199-10 T>G and novel c.1A>G mutation: two case reports and brief literature review. Medicine (Baltimore) 2017 Nov;96(45):e8549.
- 12. Carmona SM, Abacan MA, Alcausin MM. Carnitine-acylcarnitine translocase deficiency with c.199-10T>G mutation in two Filipino neonates detected through parental carrier testing. Int J Neonatal Screen 2023 Jan;9(1):4.
- 13. Van der Aa N, Zamani Esteki M, Vermeesch JR, Voet T. Preimplantation genetic diagnosis guided by single-cell genomics. Genome Med 2013 Aug;5(8):71.