Charcot-Marie-Tooth (CMT) disease is a group of inherited neurological disorders characterized by a slow progressive degeneration of peripheral nerves resulting in a loss of sensation in the limbs, fingers, and toes and weakening of the muscles in the foot, lower leg, hand, and forearm. The disease is classified into demyelinating (CMT1) or axonal (CMT2) based on electrophysiological and pathological findings.1 Inheritance of CMT can be autosomal dominant (AD), autosomal recessive (AR), or X-linked. AR demyelinating subtypes (ARCMT1 or CMT4) are earlier in onset and follow a more severe disease course than dominant CMT. Mutations in at least 10 loci have been associated with recessive demyelinating CMT.2
We describe the co-occurrence of optic neuritis and schwannoma in a patient with molecularly-confirmed CMT4B1 (CMT neuropathy type 4B1). To the best of our knowledge, these two clinical features in the same patient have never been reported with any form of CMT including CMT4B1.
Case Report
Our patient was a 28-year-old male who had completely normal motor development in the first four years of life. He then started developing bilateral, symmetrical limb weakness distally, which progressed very slowly to involve his proximal muscles. Initially able to play with other children, at around the age of nine years, he was no longer able to keep up with his peers. He was noted since early childhood to have a hoarse voice with reduced pitch. He remained able to walk and pursue other activities and had a good school performance through to university.
At the age of 20, he developed headaches and acute vision loss. He was diagnosed with optic neuritis and had a significant visual impairment that improved gradually over one year with a persistent reduction in visual acuity.
At the age of 22, he suffered a left femur fracture, after which he was unable to stand without support and became wheelchair-bound.
At the age of 27, he developed sudden-onset burning sensations all over his body below the neck. It was associated with severe and acute worsening of his motor weakness, loss of sphincter control, shortness of breath, and aphonia.
He was diagnosed with cervical cord root (C2 and C3) schwannoma, which was successfully removed surgically. Following surgery, he regained most skills lost except for some permanent motor symptoms. Currently, he is unable to use a spoon or write with a pen as before and has developed progressive chewing difficulty.
On physical examination, the patient had a low-pitched voice, bilateral facial weakness, and reduced visual acuity. Motor neurological examination revealed severe wasting of distal and proximal muscles of all limbs with hypotonia and areflexia. He had evident bilateral claw hand deformity with wrist drop in his upper extremities as well as edematous, cold, and blue-discolored feet. Distal muscle groups had no perceived movements. There was mild scoliosis with right chest wall deformity.
Investigations performed on the patient included nerve conduction velocities (NCV) with no motor or sensory potentials obtained from all nerves in the upper and lower limbs, normal magnetic resonance imaging (MRI) of brain and cervical spine, and mildly delayed visual evoked potential.
Family history revealed that the proband was of Omani Arab ethnic background with his parents being first cousins, once removed. The patient has two other male siblings with a similar motor, facial, and voice weakness [Figure 1: V:7 and V:8] and seven other healthy siblings. Five other individuals, who are double cousins, have a similar motor weakness (VI:6, VI:7, VI:8, VI:11 and VI:13). A two-year-old cousin was later diagnosed with CMT4B1 (VI:2).
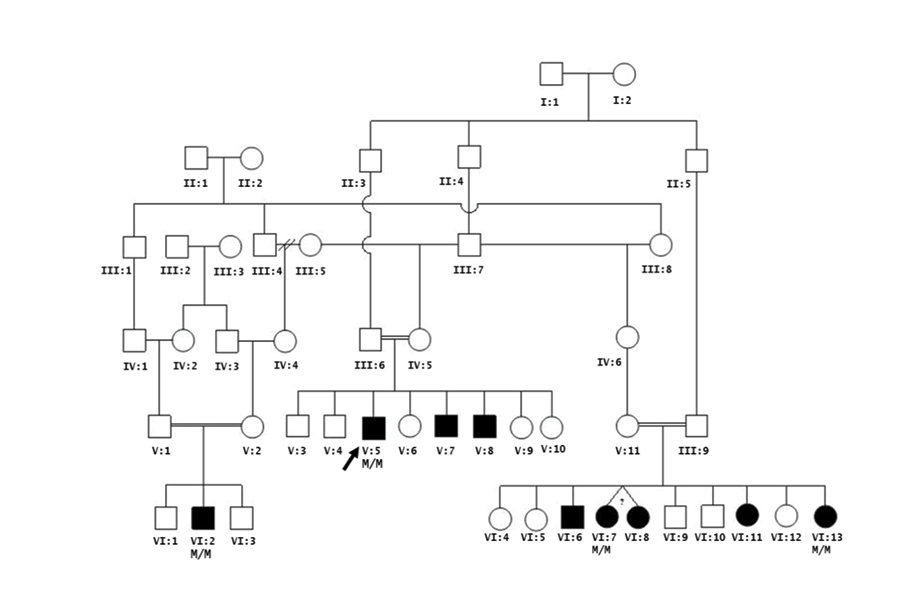 Figure 1: Family pedigree of the proband (V:5) affected with CMT4B1 with associated cervical cord schwannoma and optic neuritis. Five other individuals who are double cousins have a similar motor weakness (VI:6, VI:7, VI:8, VI:11 and VI:13). A two-year-old cousin was diagnosed with CMT4B1 (VI:2).
Figure 1: Family pedigree of the proband (V:5) affected with CMT4B1 with associated cervical cord schwannoma and optic neuritis. Five other individuals who are double cousins have a similar motor weakness (VI:6, VI:7, VI:8, VI:11 and VI:13). A two-year-old cousin was diagnosed with CMT4B1 (VI:2).
There was no history suggestive of optic neuritis or diagnosis of schwannoma in any of the other eight affected family members, although all are younger in age to the proband. All individuals in the pedigree are from the same tribe and village. Although likely, a current family record does not allow this mutation to be traced to a common ancestor. The chronologic ages in years (y) for affected individuals are as follows: V:5 28y; V:7 25y; V:8 23y; VI:2 2y; VI:6 27y; VI:7 and VI:8 26y; VI:11 23y and VI:13 15y. Genetic testing of the MTMR2 gene was initially performed on V:5, VI:2, VI:7, and VI:13. All affected individuals are confirmed homozygotes (M/M) for the c.1768C>T (p.Gln590*) MTMR2 mutation.
Bidirectional sequencing of the coding region and exon-intron boundaries of the myotubularin-related protein 2 gene (MTMR2, NG_008333.1) in the proband was performed at Diagenom, Rostock, Germany. A novel homozygous nonsense variation was identified in exon 14 of the MTMR2 gene [Figure 2]. The C>T substitution at nucleotide position 1768 (c.1768C>T) results in a premature stop codon at 590 (p.Gln590*). If translated, the protein would lack the c-terminal coiled-coil domain (aa 593-627), similar to the reported pathogenic p.Trp583* mutation.3
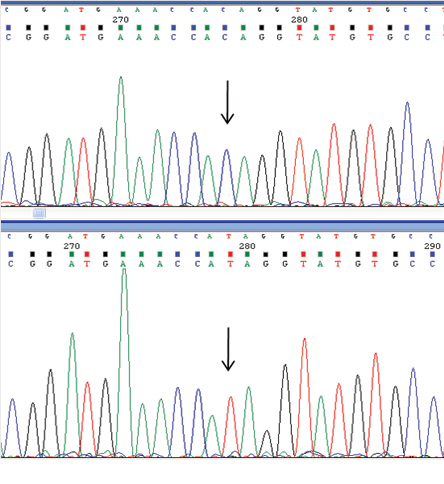 Figure 2: Electropherogram of the c.1768C>T mutation in the MTMR2 gene. This nucleotide variant is predicted to result in p.Gln590* change at the amino acid level. The arrow indicates the site of the mutation. Upper panel, wild type sequence; lower panel, mutated sequence.
Figure 2: Electropherogram of the c.1768C>T mutation in the MTMR2 gene. This nucleotide variant is predicted to result in p.Gln590* change at the amino acid level. The arrow indicates the site of the mutation. Upper panel, wild type sequence; lower panel, mutated sequence.
Genetic testing performed locally has confirmed homozygosity for the same mutation in the affected relatives as well as in a second, apparently unrelated, affected individual. Both families are from distinct geographic regions within Oman.
Discussion
Our patient had a typical CMT phenotype of severe early onset motor and sensory neuropathy with undetectable NCVs. Associated clinical features included vocal cord paresis, facial weakness, prominent chest deformities, claw hands, and a family history consistent with autosomal recessive inheritance. His clinical phenotype was consistent with CMT4B1 and molecular analysis revealed the homozygous presence of the MTMR2 mutation.
Contrasting with the classical presentation of CMT4B, our patient also had cervical cord schwannoma and optic neuritis. The individual association of either schwannoma4 or optic neuritis5 has been documented with dominant CMT1A but never in combination, nor associated with, recessive CMT. Vestibular schwannoma has also been reported with autosomal recessive CMT as a result of KARS gene mutations, but our patient had developmental delay, self-abusive behavior, and dysmorphic features.6
The absence of cervical cord schwannoma and optic neuritis in the patient’s two affected siblings and his six affected cousins suggests that MTMR2 mutations may increase the susceptibility for these two phenotypes, but they are still not frequent associations of the disease. However, this apparently increased susceptibility may be age-related as the patient was the eldest among the affected individuals in the family.
The finding of this association, as only identified in the proband, may not alter the clinical care or surveillance guidelines about the apparently increased risk for schwannoma. However, the patient was counseled about the possible association of optic neuritis and schwannoma with the underlying CMT disease and the need for urgent medical attention in the case of the development of sudden motor deterioration or focal neurological abnormalities in the proband or affected relatives. Because consanguinity is a common practice in this area, premarital genetic testing was offered to the rest of family members.
The MTMR2 gene is part of the myotubularin (MTM) family of phosphoinositide lipid phosphatases. The encoded protein possesses phosphatase activity towards phosphatidylinositol-3-phosphate and phosphatidylinositol-3,5-bisphosphate, which are both regulators of the endosomal pathway.7 CMT4B1 is the only phenotype associated with mutations in the MTMR2 gene.8 The MTMR2 coiled-coil domain predicted to be truncated as a result of the presence of the c.1768C>T mutation (p.Trp590*) in this patient, is critical for the interaction with catalytically inactive phosphatases SET binding factor 1 (SBF1)9 and SET binding factor 2 (SBF2).10 It is through this interaction that both SBF1 and SBF2 increases the enzymatic activity of MTMR2 and dictates its subcellular localization.9,11
Conclusion
This case represents the first report of the presence of schwannoma and optic neuritis in a patient with a recessive form of demyelinating neuropathy. The patient was also found to carry a novel mutation in the MTMR2 gene. In similar cases, we recommend immediate medical attention to rule out the possibility of schwannomas in patients with all demyelinating CMT subtypes in case of the development of focal neurological signs or acute worsening of clinical status. Demonstration of the mutation segregating in a second and apparently unrelated family warrants targeted mutation analysis in a symptomatic Omani patient suspected of having a recessive form of CMT.
Disclosure
The authors declared no conflicts of interest.
references
- Reilly MM, Murphy SM, Laurá M. Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2011 Mar;16(1):1-14.
- Tazir M, Bellatache M, Nouioua S, Vallat JM. Autosomal recessive Charcot-Marie-Tooth disease: from genes to phenotypes. J Peripher Nerv Syst 2013 Jun;18(2):113-129.
- Verny C, Ravisé N, Leutenegger AL, Pouplard F, Dubourg O, Tardieu S, et al. Coincidence of two genetic forms of Charcot-Marie-Tooth disease in a single family. Neurology 2004 Oct;63(8):1527-1529.
- Kwon JY, Chung KW, Park EK, Park SW, Choi BO. Charcot-Marie-Tooth 1A concurrent with schwannomas of the spinal cord and median nerve. J Korean Med Sci 2009 Aug;24(4):763-766.
- Wakerley BR, Harman FE, Altmann DM, Malik O. Charcot-Marie-Tooth disease associated with recurrent optic neuritis. J Clin Neurosci 2011 Oct;18(10):1422-1423.
- McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, et al; NISC Comparative Sequencing Program. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 2010 Oct;87(4):560-566.
- Hnia K, Vaccari I, Bolino A, Laporte J. Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends Mol Med 2012 Jun;18(6):317-327.
- Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MA, Georgiou DM, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet 2000 May;25(1):17-19.
- Kim SA, Vacratsis PO, Firestein R, Cleary ML, Dixon JE. Regulation of myotubularin-related (MTMR)2 phosphatidylinositol phosphatase by MTMR5, a catalytically inactive phosphatase. Proc Natl Acad Sci U S A 2003 Apr;100(8):4492-4497.
- Robinson FL, Dixon JE. The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13, a membrane-associated pseudophosphatase also mutated in type 4B Charcot-Marie-Tooth disease. J Biol Chem 2005 Sep;280(36):31699-31707.
- Berger P, Berger I, Schaffitzel C, Tersar K, Volkmer B, Suter U. Multi-level regulation of myotubularin-related protein-2 phosphatase activity by myotubularin-related protein-13/set-binding factor-2. Hum Mol Genet 2006 Feb;15(4):569-579.