|
Abstract
Human hydrocephalus is a disorder of abnormality in CSF flow or resorption, which has been classified in pertinent literature as congenital and acquired. Congenital hydrocephalus can present as an isolated phenomenon which is common; or with associated anomalies affecting other organs, disturbing physiology or presenting as a syndrome. This report describes a case with congenital foetal hydrocephalus, hypoplastic lungs with super-numery lobations and large left lobe of liver compared to right. Thus far, a review of the literature indicates that this case can be postulated as a subtype of Game-Friedman-Paradice syndrome.
Keywords: Congenital hydrocephalus; Supernumery pulmonary lobations; Game-Friedman-Paradice syndrome.
Introduction
Human hydrocephalus is a common medical condition that is characterized by abnormalities in the flow or resorption of cerebrospinal fluid (CSF), resulting in ventricular dilatation. It has been classified into two clinical forms, congenital and acquired.1 The development and progression of congenital hydrocephalus is a dynamic process that is not yet well understood. It is thought that it may develop at an important and specific embryonic time period of neural stem cell proliferation and differentiation in the brain. Congenital hydrocephalus may occur alone (non-syndromic) or as part of a syndrome with other anomalies (syndromic).1-7
‘Game-Friedman-Paradice syndrome’ is one of those syndromes, which is a relatively rare condition characterized by fetal growth retardation, hydrocephaly, hypoplastic multilobed lungs, and various other anomalies. The condition was observed first in four offspring from one family and reported by Game K. et al. in 1989. They postulated it to be an autosomal recessive inheritance.8
This syndrome is listed as a "rare disease" by the Office of Rare Diseases (ORD) of the National Institutes of Health (NIH). This means that Game-Friedman-Paradise syndrome, or a subtype of Game-Friedman-Paradice syndrome, affects less than one in 200,000 people in the US population.9 Unfortunately, to date, no records have been found in the Indian population as searched for.
Case Report
A 21-year-old full term, unbooked primigravida mother was brought in labor emergency in a prolonged first stage of labor. On ultrasonography, hydrocephalus was detected and the baby was found to be dead without any cardiac or cord pulsation in breech presentation. In due course, the baby was partially delivered up to the neck. Next craniotomy was performed to get the head delivered. Almost three liters of clear fluid was drained after craniotomy. Except for the macrocephaly, no gross external anomaly was detected. (Fig. 1)
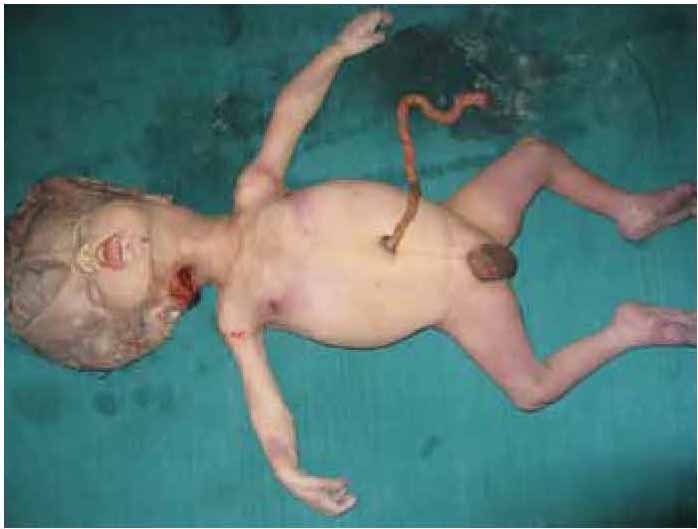
Figure 1: Macrocephalic skull could be noted and cranial diameter was contracted due to craniotomy.
Detailed history disclosed neither any records of previous medical consultation nor any antenatal ultrasonography. There was no history of exposure of any known teratogens. Both of the parents were from a Bengali population married with non-consanguinity. One of the pregnancies of the mother's aunt had fetal death with hydrocephalus, detailed investigations or postmortem examination for which was not done, as it was not reported in hospital.
Postmortem examination was carried on and documented as follows:
i) Anthropometry: body weight 2.1 kg, length 30 cm.
ii) Head: Grossly enlarged, skull with loose widely separable sutures. Skull bones were quite developed but can be easily separated. True head circumference could not be noted for post-craniotomy contracture of the diameter. (Fig. 2)
iii) Brain: Brain matter found to be too jellified to separately identify forebrain, midbrain and hindbrain. (Fig. 3)
iv) Face: Ears apparently low-set; cloudy corneae; nose, mouth, and neck normal; lower jaw small and recessed. (Fig. 2)
v) Chest:
a) Thymus was hyperplastic and grossly enlarged to cover both the lungs.
b) Heart, great vessels, and upper respiratory tract normal.
c) Right lungs had five lobes and left had three lobes. Weights were measured as 10 gm for the right and 8 gm for the left lungs (Lung weight: Body weight ratio ≤0.012) and both found to be hypoplastic.10 (Figs. 4-5)
d) On further dissection of heart, no internal anomaly found.
vi) Abdomen:
a) Left lobe of liver was grossly enlarged in comparison to right lobe. (Fig. 6)
b) There was no intestinal malposition.
c) Both the kidneys, ureters were normal.
d) The abdominal wall was developed completely.
vii) Limbs: Appeared to be normal.
viii) Spine. Normal.
ix) Perineum & groin. Normal genitalia with patent anal opening.
x) Radiographs: Normal except for enlarged cranium.
xi) Karyotyping: Normal 46.XY
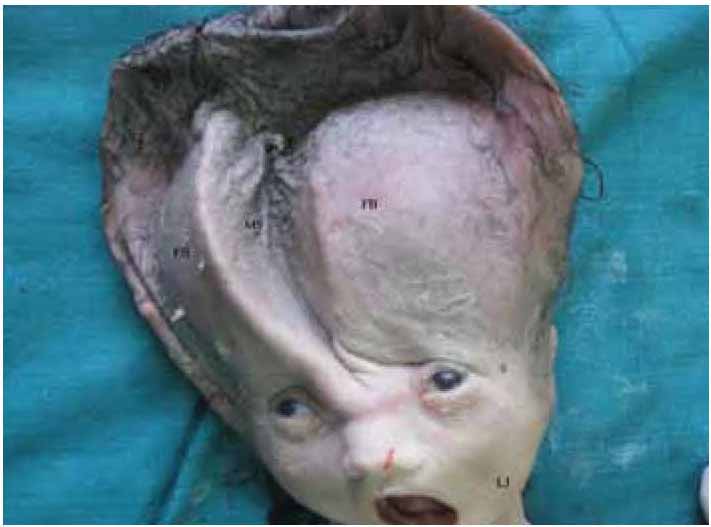
Figure 2: Showing the macrocephalic skull, wide sutures. (Metopic suture= MS), separable frontal bones (FB), recessed small jaw (LJ).
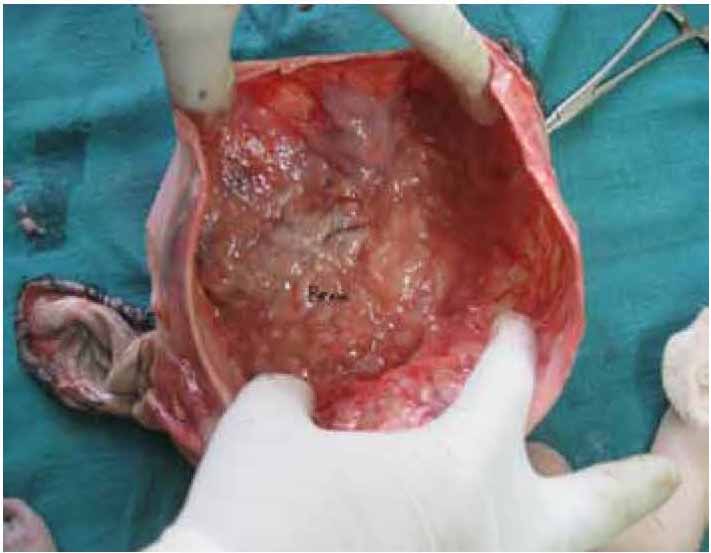
Figure 3: Jellified brain matter (BRAIN) after dissecting the skull.
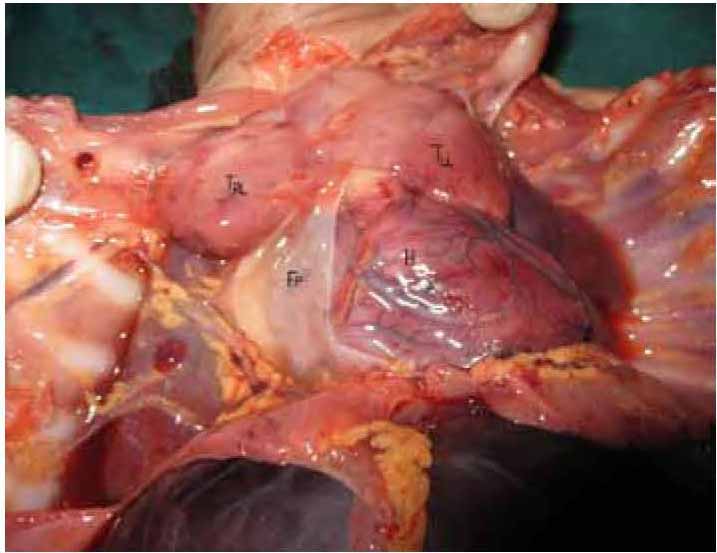
Figure 4: Enlarged thymus covering the bilateral lungs. [TRL=Right lobe of thymus, TLL=Left lobe of Thymus, H=Heart, FP=Fibrous pericardium]
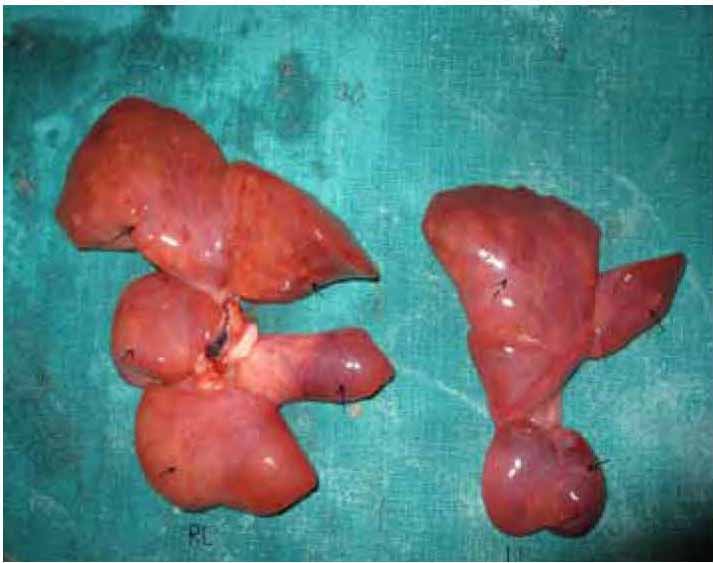
Figure 5: Right (RL) and left (LL) lungs with supernumery lobations (arrows indicating lobes).
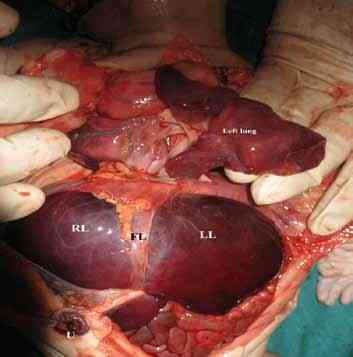
Figure 6: Huge left lobe of liver (LL) in comparison to the right lobe (RL). Falciform ligament (FL) can be well shown.
Discussion
Prominent anomalies found in this case were hydrocephalus with micrognathia, bilateral hypoplastic lungs with super-numery lobations and enlarged left lobe of liver. There were no cardiac, renal or any other skeletal anomalies. It is important to note that such trio of anomalies did not disturb the pregnancy until term and even labor was initiated in its due course. These anomalies are partial components in different combinations of Game-Paradice-Friedman syndrome,8 Hydrolethalus syndrome,11,12 Campomelic syndrome,13,14 Fullana syndrome,15 and Smith-Lemli-Opitz syndrome type-II.16,17
Typical Game-Paradice-Friedman syndrome was described by Game et al. (1989) in four siblings of a family as a combination of hydrocephalus with patent aqueduct of sylvius, hypoplastic lungs with extra lobation, intestinal malrotation with or without omphalocoele and skeletal anomalies.8 Here neither intestinal malrotation nor any skeletal anomalies were found.
On the contrary, cardiac and palate malformation are compulsory components of other four syndromes mentioned earlier. Again Mirfazeli et al. (2012), in their study have showed quiet a number of patients with cleft lip had associated hydrocephalus, but still the complete range of anomalies viz. lung multilobation and visceral anomalies was not demonstrated.18 Associated genito-renal anomaly is compulsory for Campomelic syndrome, Fullana syndrome as well as Smith-Lemli-Opitz syndrome. Comparing the present case, maximum similarities were found with Game -Friedman-Paradice syndrome, though some of its features are not present here. Patency of aqueduct of Sylvius could not be demonstrated as the brain matter was grossly liquified. (Table 1)
Over and above history of similar fetal death in maternal ancestry also indicates it to be an autosomal recessive disorder like Game-Friedman-Paradice syndrome. Any subtype of Game-Friedman-Paradice syndrome to date awaits to be demonstrated clearly in the literature so far searched for; but still, this case can be considered as its partial presentation.
Table 1: A comparison chart showing the compulsory components of the syndromes described here along with the present case.
Uncovered detailed history of similar fetal death in maternal family due to non-reporting in hospital and undone further genetic investigations besides the basic karyotyping and radiological investigations like MRI, due to infrastructural limitation may be considered as limitation of this study.
Acknowledgements
Authors are grateful to HOD and faculties of Department of Obs-Gynae, North Bengal Medical College and especial thanks to Dr. Tapan Mukerji PhD, Associate Professor in Rock Physics (Geology) of San Fransico University, USA; for their help.
References
1. Mori K, Shimada J, Kurisaka M, Sato K, Watanabe K. Classification of hydrocephalus and outcome of treatment. Brain Dev 1995 Sep-Oct;17(5):338-348.
2. Mori K. Hydrocephalus–revision of its definition and classification with special reference to "intractable infantile hydrocephalus". Childs Nerv Syst 1990 Jun;6(4):198-204.
3. Haverkamp F, Wölfle J, Aretz M, Krämer A, Höhmann B, Fahnenstich H, et al. Congenital hydrocephalus internus and aqueduct stenosis: aetiology and implications for genetic counselling. Eur J Pediatr 1999 Jun;158(6):474-478.
4. Adams C, Johnston WP, Nevin NC. Family study of congenital hydrocephalus. Dev Med Child Neurol 1982 Aug;24(4):493-498.
5. Fernell E, Hagberg B, Hagberg G, von Wendt L. Epidemiology of infantile hydrocephalus in Sweden. I. Birth prevalence and general data. Acta Paediatr Scand 1986 Nov;75(6):975-981.
6. Teebi AS, Naguib KK. Autosomal recessive nonsyndromal hydrocephalus. Am J Med Genet 1988 Oct;31(2):467-470.
7. Partington MD. Congenital hydrocephalus. Neurosurg Clin N Am 2001 Oct;12(4):737-742, ix.
8. Game K, Friedman JM, Paradice B, Norman MG. Fetal growth retardation, hydrocephalus, hypoplastic multilobed lungs, and other anomalies in 4 sibs. Am J Med Genet 1989 Jun;33(2):276-279.
9. Game Friedman Paradice syndrome. Genetic and rare diseases information centre. Office Of The Rare Diseases & Research. National Institute of Health. Available at: http://rarediseases.info.nih.gov/GARD/Condition/2427/Game_Friedman_Paradice_syndrome.aspx. (Accessed on 29 February 2012).
10. Askenazi SS, Perlman M. Pulmonary hypoplasia: lung weight and radial alveolar count as criteria of diagnosis. Arch Dis Child 1979 Aug;54(8):614-618.
11. Salonen R, Herva R, Norio R. The hydrolethalus syndrome: delineation of a "new", lethal malformation syndrome based on 28 patients. Clin Genet 1981 May;19(5):321-330.
12. Toriello HV, Bauserman SC. Bilateral pulmonary agenesis: association with the hydrolethalus syndrome and review of the literature from a developmental field perspective. Am J Med Genet 1985 May;21(1):93-103.
13. Hall BD, Spranger JW. Campomelic dysplasia. Further elucidation of a distinct entity. Am J Dis Child 1980 Mar;134(3):285-289.
14. Houston CS, Opitz JM, Spranger JW, Macpherson RI, Reed MH, Gilbert EF, et al. The campomelic syndrome: review, report of 17 cases, and follow-up on the currently 17-year-old boy first reported by Maroteaux et al in 1971. Am J Med Genet 1983 May;15(1):3-28.
15. Fullana A, Garcia-Frias E, Martinez-Frias ML, Razquin S, Quero J. Caudal deficiency and asplenia anomalies in sibs. Am J Med Genet 2005;25(Suppl 2).
16. Curry CJ, Carey JC, Holland JS, Chopra D, Fineman R, Golabi M, et al. Smith-Lemli-Opitz syndrome-type II: multiple congenital anomalies with male pseudohermaphroditism and frequent early lethality. Am J Med Genet 1987 Jan;26(1):45-57.
17. Rutledge JC, Friedman JM, Harrod MJ, Currarino G, Wright CG, Pinckney L, et al. "new" lethal multiple congenital anomaly syndrome: Joint contractures, cerebellar hypoplasia, renal hypoplasia, urogenital anomalies, tongue cysts, limb shortness, eye abnormalities, defects of the heart, gallbladder agenesis, and ear malformations. Am J Med Genet 2005;19(2).
18. Mirfazeli A, Kaviany N, Hosseinpour KR, Golalipour MJ. Incidence of cleft lip and palate in gorgan - northern iran: an epidemiological study. Oman Med J 2012 Nov;27(6):461-464.
|