| |
Abstract
Skeletal dysplasia is an uncommon cause of short stature in children. An 11-year-old girl was evaluated for severe short stature in a tertiarycare hospital. Clinical examination revealed severe disproportionate short stature and classical triad of multiple supernumerary teeth, and complete absence of clavicles and open sagittal sutures and fontanelles. Skeletal survey confirmed these findings, in addition toother features associated with the syndrome.
Keywords: Short stature; Skeletal dysplasia; Cleidocranial dysplasia.
Introduction
Cleidocranial dysplasia (CCD) is a rare disorder with autosomal dominant inheritance, though 40% cases occur spontaneously with no apparent genetic cause.1 The incidence of the disorder has been reported to be 1:1000,000.2 This syndrome consists of a myriad of skeletal manifestations and short stature. Skeletal manifestations include hypoplastic or aplastic clavicle, patent sutures and fontanellae, delayed eruption of teeth, supernumerary teeth, open pubic symphysis and other skeletal abnormalities.3 This disease usually has a wide range of presentations ranging from subtle skeletal abnormalities to severe cord involvement leading to syringomyelia.4,5 The disease gene has been mapped to chromosome 6p21 within a region containing core-binding factor subunit alpha-1, a member of the runt family of transcription factors Runt-related transcription factor 2.6 This case report presents, an 11-year-old girl with severe short stature with all the features of CCD including a rare finding of complete absence of clavicles.
Case Report
An 11-year-old female child was referred to the Department of Endocrinology, SKIMS, India, for evaluation of short stature. She was a product of non-consangious marriage; first in birth order; born at term by vaginal delivery conducted at home. The patient had no significant perinatal history and had normal developmental and mental mile stones. On examination, the patient had severe disproportionate short stature with height of 118 cm (height SDS of -4.06), upper segment to lower segment ratio of 1.03 (Fig. 1), head circumference was 53 cm, weight of 18 kg, and BMI of 13.1 kg/m2. The patient was prepubertal. There was frontal and parietal bossing, open anterior fontanelle, hypertelorism, depressed nasal bridge, high arched palate, supernumerary teeth, and clinodactyly. She also had bilaterally absent clavicles, bilaterally hypermobile shoulders which could be approximated in the midline, (Fig. 1). Cardiovascular, respiratory, abdominal, and central nervous system examination was unrevealing. The patient had two more siblings who had normal height percentiles and no skeletal abnormality.
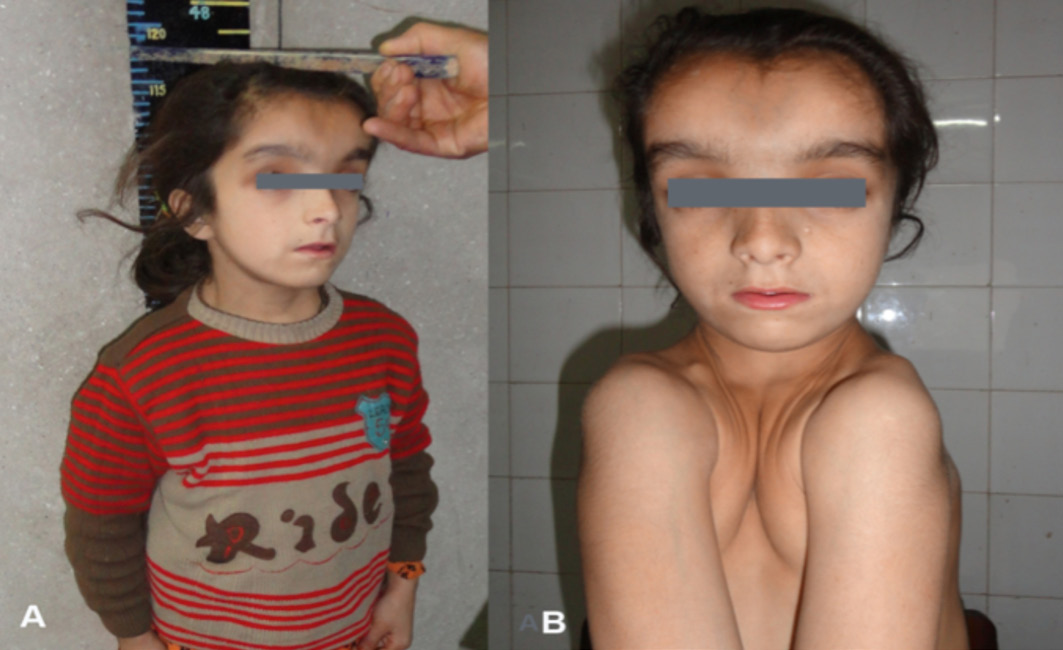
Figure 1: A) Severe short stature (height: 118 cm); B) Extreme hyperadduction of shoulders almost approximated in midline.
Laboratory investigations revealed normal hemogram with hemoglobin of 13.7 g/dL, platelets of 220 lac/mm3 and leukocyte count of 6000/mm3. Renal, liver and bone function tests including serum calcium (9.9 mg/dL), phosphorus (3.9 mg/dL) and alkaline phosphatase of (246 U/L) were normal. Ph and serum electrolytes were normal and thyroid function tests revealed TSH of 2.3 mIU/L and T4 of 9.9 µg/dL. A-12 lead electrocardiogram showed normal sinus rhythm. Ultrasound abdomen and echocardiography were normal.
X-ray left wrist AP view revealed normal bone age commensurate with her chronological age and confirmed bilateral clinodactyly, (Fig. 2). X-ray skull revealed frontal and parietal bossing, open sutures and fontanelle. There was a presence of supernumerary teeth and malocclusion of teeth, (Fig. 2). X-ray chest revealed bilateral absent clavicles and X-ray pelvis showed wide open pubic symphysis, (Fig. 3). The differential diagnosis in this patient included hypothyroidism, rickets, pyknodysostosis, hypophosphatesia, osteogenesis imperfecta, Russell-Silver syndrome, Down's syndrome, and Apert syndrome. These were easily ruled out due to the presence of classical features of CCD and normal hormonal, biochemical, echocardiographic and ultrasonographic results, in addition to the presence of normal IQ.
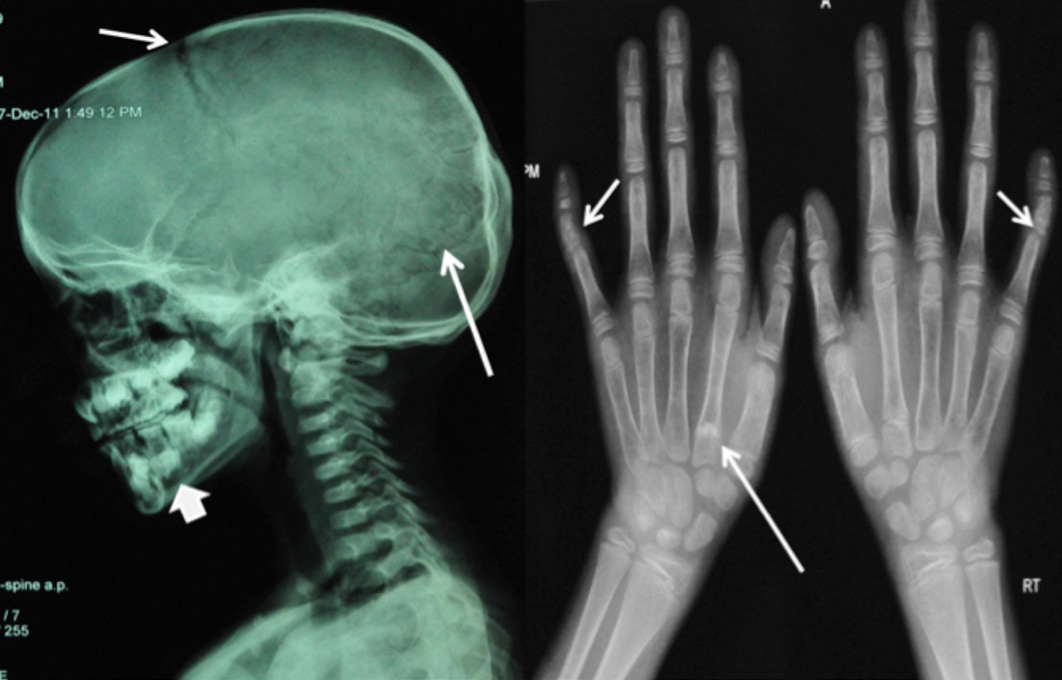
Figure 2: A) X-ray skull lateral view showing open anterior fontanella (short arrow), open cranial sutures (long arrow), frontal and parietal bossing and supernumerary teeth (arrow head); B) X-ray both hands AP view showing normal bone age and bilateral clinodyctyly (short arrow), small 2nd and 5th middle phalanx, proximal epiphyses of 2nd metacarpal bilaterally (long arrow).
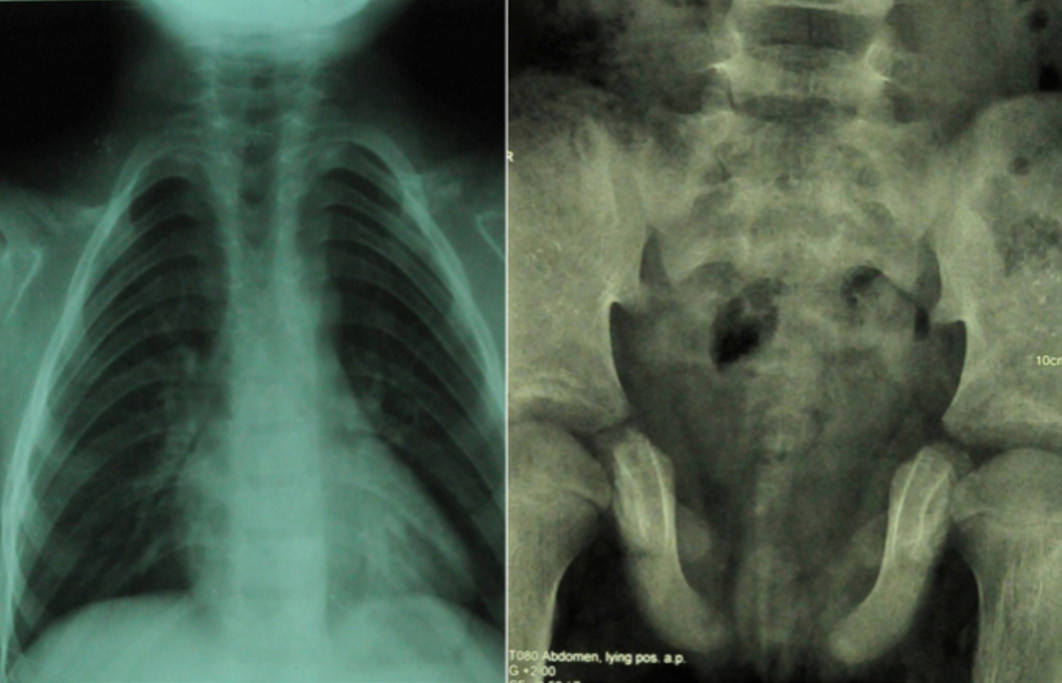
Figure 3: A) X-ray chest revealing bilateral absent clavicle, cylinder shaped chest; B) X-ray pelvis revealing wide open pubic symphysis and poorly developed femur head and neck.
Discussion
Cleidocranial dysplasia is a rare syndrome usually having autosomal dominant inheritance. Until 2009, around 700 cases had been described since its original description.7 The disorder is caused by a mutation of the core binding factor-α1 located at chromosome 6p21. The gene encodes a protein necessary for correct function of osteoblasts.4,8 Although the usual pattern of inheritance is autosomal dominant, around 40% of cases can occur as sporadic.9,10 Reports of clavicular defects appeared as early as 1765, but Scheuthauer was probably first to describe this syndrome accurately.8 The main features of CCD include clavicular hypoplasia/aplasia, open sutures and fontanelle, frontal and parietal bossing, supernumerary teeth and open pubic symphasis. The patient in this report had all the classical features of the syndrome; in addition, she had complete absence of both clavicles resulting in the classical ability of anterior approximation of both shoulders. Bilateral complete absence of clavicles is seen in a small percentage (10%) of patients with CCD, while hypoplasia of the acromial end is common.11-13 Other less common clavicular involvement include occurrence of two separate fragments of clavicle, absence of sternal end while the acromial end is present. Bilateralism is usually the rule but not always the case. The thoracic cage is small and bell shaped with short oblique ribs. Other features of the syndrome include short stature, abnormality of phalyngeal bones, maxillary and mandibular abnormalities, obliterated maxillary sinuses, crossed renal ectopia, and pelvic abnormalities.7 The widened pubic symphysis as was seen in this case is due to delayed ossification.
Other pelvic abnormalities include hypoplasia, anterior rotation of iliac wings, and wide sacroiliac joints. The dysplastic pelvis often necessitates cesarean section in pregnant females.14 A constant feature is the presence of both proximal and distal epiphyses of the 2nd metacarpal and metatarsal bones leading to excessive lengthening of these bones.11,15 This classical feature was also presented here. Other features which were present in this case included hypoplastic 2nd and 5th middle phalanx with bilateral clinodactyly, (Fig. 2). Final height is significantly reduced in cases with CCD, while the birth length is usually normal but height percentiles fall below 2 SD around age 4-8 years.16 Patients usually have disproportionate short stature with limbs shorter than the trunk. The presented case also had severe disproportionate short stature with US/LS of 1.03 and height of 118 cm (SDS of -4.2). High arched palate and dental abnormalities are other main features of CCD. This patient also had supernumerary teeth which were impeding the development of normal permanent teeth. It has therefore been suggested that supernumerary teeth should be removed as early as possible.14
Conclusion
In summary, CCD is an uncommon cause of severe short stature. A thorough history and physical examination with the support of simple radiology will help to arrive at the diagnosis in most cases. Except for severe short stature, the puberty and fertility is normal in most of the patients with CCD.
Acknowledgements
The authors reported no conflict of interest and no funding was received in this work.
References
1. Tanaka JL, Ono E, Filho EM, Castilho JC, Moraes LC, Moraes ME. Cleidocranial dysplasia: importance of radiographic images in diagnosis of the condition. J Oral Sci 2006 Sep;48(3):161-166.
2. Garg RK, Agrawal P. Clinical spectrum of cleidocranial dysplasia: a case report. Cases J 2008;1(1):377.
3. Kalliala E, Taskinen PJ. Cleidocranial dysostosis. Report of six typical cases and one atypical case. Oral Surg Oral Med Oral Pathol 1962 Jul;15:808-822.
4. Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 1999 Mar;36(3):177-182.
5. Vari R, Puca A, Meglio M. Cleidocranial dysplasia and syringomyelia. Case report. J Neurosurg Sci 1996 Jun;40(2):125-128.
6. Yoshida T, Kanegane H, Osato M, Yanagida M, Miyawaki T, Ito Y, et al. Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations. Am J Hum Genet 2002 Oct;71(4):724-738.
7. Suresh SS. A family with cleidocranial dysplasia and crossed ectopic kidney in one child. Acta Orthop Belg 2009 Aug;75(4):521-527.
8. Zhou G, Chen Y, Zhou L, Thirunavukkarasu K, Hecht J, Chitayat D, et al. CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia. Hum Mol Genet 1999 Nov;8(12):2311-2316.
9. McNamara CM, O’Riordan BC, Blake M, Sandy JR. Cleidocranial dysplasia: radiological appearances on dental panoramic radiography. Dentomaxillofac Radiol 1999 Mar;28(2):89-97.
10. Li Y, Pan W, Xu W, He N, Chen X, Liu H, et al. RUNX2 mutations in Chinese patients with cleidocranial dysplasia. Mutagenesis 2009 Sep;24(5):425-431.
11. Jarvis JL, Keats TE. Cleidocranial dysostosis. A review of 40 new cases. Am J Roentgenol Radium Ther Nucl Med 1974 May;121(1):5-16.
12. Mehta DN, Vachhani RV, Patel MB. Cleidocranial dysplasia: a report of two cases. J Indian Soc Pedod Prev Dent 2011 Jul-Sep;29(3):251-254.
13. Karagüzel G, Aktürk FA, Okur E, Gümele HR, Gedik Y, Okten A. Cleidocranial dysplasia: a case report. J Clin Res Pediatr Endocrinol 2010;2(3):134-136.
14. Cooper SC, Flaitz CM, Johnston DA, Lee B, Hecht JT. A natural history of cleidocranial dysplasia. Am J Med Genet 2001 Nov;104(1):1-6.
15. Jensen BL. Somatic development in cleidocranial dysplasia. Am J Med Genet 1990 Jan;35(1):69-74.
16. Levin EJ, Sonnenschein H. Cleidocranial dysostosis. N Y State J Med 1963 May;63:1562-1566. Sonnenschein H.
|
|