Chronic obstructive pulmonary disease (COPD) refers to a group of lung diseases characterized by airway inflammation and tissue destruction with non-reversible airflow limitation. Airflow limitation is generally progressive and associated with an abnormal response of the lungs to noxious particles or gases such as cigarette smoke, coal mining dust, diesel exhaust particles, and fumes from burning biomass fuels for cooking or heating that leads to chronic airway inflammation.1 There is a broad range of inflammatory cells like macrophages, neutrophils, T and B lymphocytes, eosinophils, and epithelial cells involved in the inflammatory process of COPD.2 Today COPD is acknowledged as a systemic disease that not only affects the airways and lungs.3 Neutrophils are one of the main effector cells in COPD and during exacerbations, which are frequently caused by bacterial infections. Neutrophils are rapidly recruited to the site of action and play a significant role in tissue damage and killing bacteria.4,5
Neutrophils, together with other innate immune cells, are involved in inflammation, interact with pathogens and/or damage associated molecular patterns (PAMPs and/or DAMPs). These molecular patterns are sensed through highly conserved pattern-recognition receptors (PRRs), called Toll-like receptors (TLRs). TLR2 recognizes lipoproteins from Gram-positive bacteria with other wide range of components like glycopeptides, peptidoglycan, and lipoarabinomannan.6 TLR4 recognizes lipoproteins from Gram-negative bacteria and binds lipopolysaccharide (LPS) and lipooligosaccharide (LOS).7,8 A signaling cascade is initiated upon activation of PRRs that lead to the activation of nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-kB), which up-regulate the production of inflammatory mediators, chemokines, and cytokines.9,10 Both TLR2 and TLR4 are widely studied and appear to have both endogenous and exogenous ligands. It has been shown that patients with COPD express higher TLR2 and TLR4 than healthy controls from airway samples,11 but in peripheral blood neutrophils, these receptors have been lesser studied. TLR2 and TLR4 are believed to have a regulating role as a snare receptor in host innate immune response against pathogens. Therefore, it results in a negative regulation of cytokines and chemokines.
Environmental exposures are also triggering the innate immune response in COPD patients. A persistent innate immune activation is associated with COPD.12 This persistent immune activation is concerned to the interaction of PRRs with pathogens, reactive oxygen species, and damaged cells, which lead to the development and exacerbations in COPD.13,14 There is an imbalance between proteases and antiproteases which leads to excessive proteinase activity that can cause host tissue damage in COPD.15 Neutrophils and macrophages are the major source of matrix mettaloproteinase-9 (MMP-9) and neutrophil elastase (NE). Neutrophils get activated and release proteinase (including NE, MMP-9 and MMP-8), which have been shown to be increased in COPD.16 The neutrophil cells are also associated with interleukin-8 (IL-8) mediated neutrophilic inflammatory responses in the airways of COPD patients.17 Altered activities of neutrophilic cells are found in the peripheral blood of patients with stable COPD and upregulation of inflammatory genes18 with increased respiratory burst during exacerbations.19
Our study aimed to analyze the expression of innate immune receptors in peripheral blood neutrophils of patients with COPD compared to their healthy counterparts. However, there is no proper predictive marker for the early diagnosis of airway obstructions in patients with COPD. Therefore, we examined the innate immune response in circulating neutrophils stimulated with TLR 2/4 agonist, LPS, and associated expression of IL-8 and MMP-9 while controlling for the effect of smoking, and the presence and severity of airway obstruction. To examine these effects, we also assessed the relationship between smoking status and airflow obstruction.
Methods
This study was conducted at the Department of Respiratory Medicine, King George’s Medical University, Lucknow, India and was approved by the ethics committee of the institution. All participants gave their written informed consent before inclusion in this study. We enrolled (n = 101) patients with COPD who were referred to our respiratory disease outpatient clinic and healthy controls (n = 101) having forced expiratory volume in the first second (FEV1) > 80% of predicted value and without any other respiratory disease in the study. All the patients were diagnosed according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD).1 Patients with COPD had to be in a stable state (no exacerbations in the last six weeks) for inclusion into the study. Patients with confirmed history of smoking and a FEV1/forced vital capacity (FVC) ratio of < 70% after salbutamol administration were enrolled in the study. Inclusion criteria for the healthy control group were the absence of COPD confirmed by history, physical evaluation, and spirometry. Patients were excluded if they had any other respiratory disease, acute infections, and other inflammatory diseases. Demographic characteristics, clinical assessment, medical history (including smoking status) of the studied participants were recorded. Peripheral blood samples were obtained from all participants.
Pulmonary function tests were performed using pulmonary function equipment (Cosmed, Italy) by a single technician. After three consecutive tests, the best test was accepted. According to American Thoracic Society guidelines, FEV1, FVC, and FEV1/FVC were measured and COPD staging was done according to GOLD 2013 criteria.1
Peripheral blood neutrophils isolation was performed as per methodology previously used by Clark and Nauseef.20 Neutrophil cells were resuspended in RPMI 1640 (Gibco Invitrogen, USA) supplemented with 10 mM HEPES, 1% fetal calf serum and 1% antibiotics (penicillin/streptomycin). Neutrophil cells were cultured at 1 × 106 cells/mL ± LPS (100 ng/mL Escherichia coli LPS, Sigma, USA) at 37 oC (5% CO2) for 24 hours. Cell-free supernatants were prepared for enzyme-linked immunosorbent assay (ELISA) and cell pellets stored in RLT buffer (Qiagen, Germany) at -80 oC for RNA extraction.
Table 1: Primers used for Toll-like receptor 2 (TLR2), TLR4, and ß-actin.
|
TLR2 |
5’-TGCTTTCCTGCTGGAGATTT-3’ |
5’-TGTAACGCAACAGCTTCAGG-3’ |
|
TLR4 |
5’-TTCAAGACCAAGCCTTTCAG-3’ |
5’-CATAGTCCTTCCATGATAGA-3’ |
Total RNA was extracted from peripheral blood neutrophils stimulated by LPS using a commercially available RNA isolation kit (RNeasy Mini kit, Qiagen, Hilden, Germany), according to the manufacturer’s instructions. After purification of RNA we performed DNase I treatment to assure highly pure RNA without genomic DNA contamination. The quality and quantity of extracted RNA was determined by nanodrop. High capacity cDNA Reverse Transcription kit (Applied Biosystems Foster city CA, USA) was used reverse transcribed to cDNA from 100 ng of RNA. Real-time polymerase chain reaction (PCR) was performed by ABI 7500 real-time PCR machine (Applied Biosystem USA) using an ABI Power SYBR Green Master mix (Applied Biosystems, USA). The primers used for TLR2, TLR4, and ß-actin were shown in Table 1. The results were calculated using 2-Δct method normalizing to the internal calibrator Beta-actin.
IL-8 and MMP-9 levels were determined in cell culture supernatant of peripheral blood neutrophils stimulated by LPS using the ELISA technique according to the manufacturer instructions (Ray Bio, Human IL-8/MMP-9 ELISA Kit, Norcross, USA).
Data were analyzed using GraphPad Prism version 5 (GraphPad software Inc.; La, Jolla, CA, USA). All data were expressed as mean ± standard error of the mean (SEM). The chi-square test was used for categorical data and groups were compared by unpaired t-test or one-way analysis of variance (ANOVA). The Bonferroni test was applied for multiple comparisons. Spearman’s rank correlation tests were used for determining associations between data. Data with p < 0.050 were considered significant.
Results
Patients with moderate (n = 15), severe (n = 65) and very severe (n = 21) COPD were enrolled in the study according to the GOLD criteria. The demographic and clinical characteristics of all the participants are shown in Table 2. The mean age of patients in the COPD and control group were not significantly different (p = 0.070). The male/female sex ratio of COPD patients was higher compared to the healthy control group (p = 0.008). A significant difference was found in smoking history and pack years smoked (p = 0.001). In the control group, it was 18.7±1.3 pack years and 32.8±1.8 pack years in the COPD group. COPD patients had significantly lower lung function compared to healthy controls. The pulmonary function tests of patients in the COPD group showed predicted mean post-bronchodilator FEV1 of 37.6±1.0% and a predicted FVC of 59.7±1.4% with a mean FEV1/FVC ratio of 61.2±0.7%. The healthy controls showed FEV1 of 89.8±0.7% and FVC 88.7±0.9% with a mean FEV1/FVC ratio of 101.6±0.7%.
The gene expression of TLR2 on peripheral blood neutrophils was significantly higher in the COPD group compared to the control group [Figure 1a; p = 0.012]. Similarly, TLR4 gene expression was also increased in the COPD group compared to the control group [Figure 1b; p = 0.015]. Patients in the COPD group had significantly higher levels of IL-8 [Figure 1c; p = 0.004] and MMP-9 [Figure 1d; p = 0.010] protein than patients in the control group.
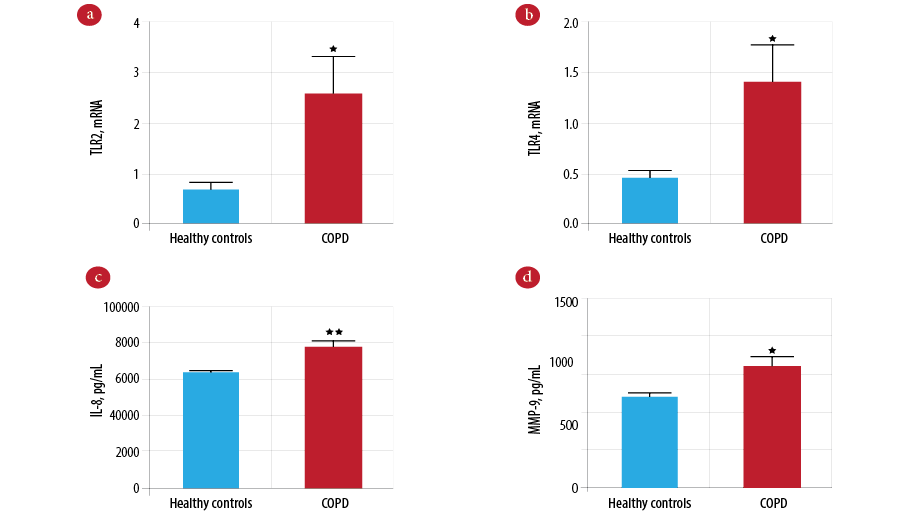
Figure 1: Comparison of innate immune mediators between the chronic obstructive pulmonary disease (COPD) and control groups. Gene expression of Toll-like receptor 2 (TLR2) (a) and Toll-like receptor 4 (TLR4) (b) measured by real-time polymerase chain reaction (PCR) in COPD (n = 101) and controls (n = 101). Protein level of interleukin-8 (IL-8) (c) and mettaloproteinase-9 (MMP-9) (d) measured by enzyme-linked immunosorbent assay (ELISA) in COPD (n = 101) and controls (n = 101). Data presented as mean± standard error of the mean (SEM) and compared by unpaired t-test. *p-values < 0.050 are considered significant. **p-values <0.010 vs. the healthy control group.
Table 2: Clinical characteristics of patients in the COPD and healthy control groups.
|
Age, mean ± SEM, years |
52.9 ± 1.1 |
55.6 ± 0.9 |
0.070a |
|
Gender, M/F |
69/32 |
85/16 |
0.008b |
|
Height, mean ± SEM, cm |
161.1 ± 0.9 |
160.2 ± 0.9 |
0.460a |
|
Weight, mean ± SEM, kg |
62.8 ± 1.3 |
50.6 ± 1.0 |
0.001a |
|
BMI, mean ± SEM, kg/m2 |
24.1 ± 0.4 |
19.8 ± 0.4 |
0.001a |
|
Smoking, never/smoker |
68/33 |
28/73 |
0.001b |
|
Pack years, mean ± SEM |
18.7 ± 1.3 |
32.8 ± 1.8 |
0.001a |
|
Post FVC, mean ± SEM, % |
88.7 ± 0.9 |
59.7 ± 1.4 |
0.001a |
|
Post FEV1 predicted, mean ± SEM, % |
89.8 ± 0.7 |
37.6 ± 1.0 |
0.001a |
Data were expressed in mean ± standard error of the mean (SEM). p < 0.050 was considered significant.
aUnpaired t-test, bchi-square test; FVC: forced vital capacity; BMI: body mass index; FEV1: forced expiratory volume in the first second; COPD: chronic obstructive pulmonary disease.
The effect of smoking was investigated by further analysis of innate immune mediators in smokers and nonsmokers in both the COPD and control groups. Patients with COPD that smoked had significantly higher TLR2 gene expression [Figure 2a; p = 0.047] and TLR4 [Figure 2b; p = 0.012] and significantly increased levels of IL-8 [Figure 2c; p = 0.016] and MMP-9 [Figure 2d; p = 0.043] compared to smokers in the control group and all nonsmokers. However, there was a strong correlation observed between the smoking history (pack years) with IL-8 marker and measures of airflow obstructions. Smoking pack years was significantly positively correlated with IL-8 levels (p = 0.002) and negatively correlated with FEV1% predicted (p = 0.023) and FEV1/FVC (p = 0.011) [Table 3].
To examine the effect of severity in COPD, we compared patients with very severe (n = 21) and severe COPD (n = 65) to patients with moderate COPD (n = 15). We found the gene expression of TLR2 was higher in very severe COPD, but this was not statistically different from severe to moderate COPD patients [Figure 3a; p = 0.354]. Reduced expression of TLR4 gene was found in very severe COPD patients [Figure 3b; p = 0.041]. There was a significantly elevated level of IL-8 in very severe COPD patients compared to severe and moderate COPD patients [Figure 3c; p = 0.026]. However, increased levels of MMP-9 in very severe COPD patients was not statistically significant [Figure 3d; p = 0.058].
Discussion
Inflammation and inflammatory mediators are important components of occurrence and progression of COPD. Neutrophils are the primary component of the innate immune response and play a key role in the COPD-associated inflammatory process. In this study, circulating neutrophils was isolated from patients with COPD and healthy controls and the role of TLR2, TLR4, IL-8, and MMP-9, in the context of smoking and COPD severity, was evaluated. The increased expression of TLR2/4 in circulating neutrophils of COPD indicated chronic activation of innate immune responses. Similarly, the increased levels of IL-8 and MMP-9 in COPD patients may suggest that the COPD is an inflammatory condition [Figure 1].
Furthermore, we also observed a correlation of smoke pack years with inflammatory markers and pulmonary function in terms of FEV1% and FEV1/FVC. There is an association between smoking and more inflammation in COPD when these patients were compared with nonsmokers. The level of all the studied inflammatory markers is higher in smokers with COPD. Further, smoke pack years is positively correlated with IL-8 levels and negatively correlated with FEV1% and FEV1/FVC. This results support that the inflammatory neutrophil granulocyte is one cause of COPD and smoking is driving this factor [Figure 2 and Table 3]. We also found an association between the severity of COPD and inflammation. However, this was not statically significant, possibly due to the small sample size [Figure 3].
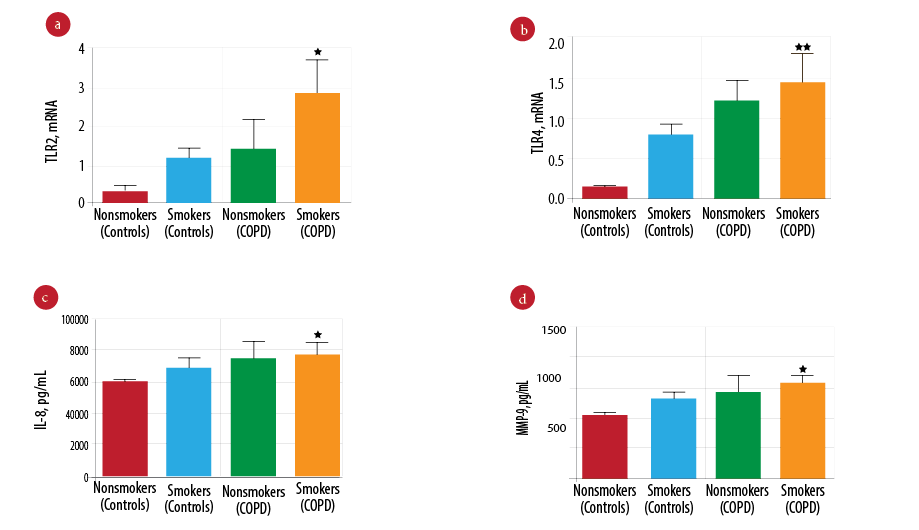
Figure 2: Comparison of innate immune mediators between patients in the chronic obstructive pulmonary disease (COPD) (smokers and nonsmokers) and control groups (smoker and nonsmokers). Gene expression of Toll-like receptor 2 (TLR2) (a) and Toll-like receptor 4 (TLR4) (b) measured by real-time polymerase chain reaction (PCR) in COPD (smokers (n = 73), nonsmokers (n = 28)) and control group (smokers (n = 33) and nonsmokers (n = 68)). Protein level of interleukin-8 (IL-8) (c) and mettaloproteinase-9 (MMP-9) (d) measured by enzyme-linked immunosorbent assay (ELISA) in COPD (smokers (n = 73) and nonsmokers (n = 28)) and control group (smokers (n = 33) and nonsmokers (n = 68)). Data presented as mean± standard error of the mean (SEM) and compared by one-way analysis of variance (ANOVA) test followed by Boneferroni post-test for multiple comparisons. **p-values < 0.010 vs. the nonsmokers control group. *p-values < 0.050 vs. the nonsmokers control group.
Cigarette smoking is a known risk factor for COPD and our study exhibits a greater history of smoking in patients with COPD [Table 2]. Cigarette smoke induces an inflammatory response that includes macrophage, neutrophil, monocytes, T-lymphocyte attracting factor, proinflammatory mediators, and proteolytic enzymes.21,22 Many previously conducted studies have suggested the importance of upregulated cytokine and inflammatory genes; also establishing the migration and cytotoxic responses of systemic neutrophils in COPD.11,18,19,23–25 Neutrophils may release an extensive quantity of IL-8, which adds to the positive feedback circle in COPD. We observed that the level of IL-8 was higher in smokers with COPD compared to nonsmoking healthy controls. We also observed a significant increase in the level of IL-8 with increasing COPD severity. The results of the current study are consistent with previous studies in serum and bronchoalveolar fluids from patients with mild, moderate, and severe COPD.26,27 They found that IL-8 levels increased with increasing severity of COPD. An increased level of IL-8 in peripheral blood neutrophils of COPD patients suggests a systemic inflammatory effect on these patients.
MMP-9 is one of the most important MMP in its family and is responsible for tissue repair and remodeling.28,29 The increased level of MMP-9 may significantly increase the elastolytic load in the lungs, which accelerate the loss of lung functions. An Egyptian study demonstrated that MMP-9 level was increased in patients with COPD and smokers in the healthy control group compared with healthy nonsmokers.30 Similarly, another study showed the levels of MMP-9 were significantly higher in COPD and the levels were increasing in severe to very severe stages.31 Our study showed MMP-9 levels were significantly higher in smokers with COPD compared to nonsmoking healthy controls. MMP-9 levels also increase in very severe stages of COPD compared to patients with severe and moderate forms of the disease. However, the difference was not statically significant between these groups. This increased level of MMP-9 may lead to the destruction of the extracellular matrix in airways and may contribute to decline the lung function of COPD.
Table 3: Correlation between pack years, IL-8 level, and spirometry parameters in peripheral blood neutrophils of patients with COPD.
|
IL-8 level |
0.44 |
0.002 |
|
Post FEV1% |
-0.33 |
0.023 |
IL-8: interleukin 8; COPD: chronic obstructive pulmonary disease; FEV1: forced expiratory volume in the first second; FVC: forced vital capacity. Coefficient of correlation. p < 0.050 was considered significant.
The role of TLRs in COPD pathogenesis is an emergent interest, along with its association between altered expression of TLRs and cigarette smoke exposure.32 TLR2 and TLR4 upon activation, activate the NF-kB pathway that regulates neutrophil activation, migration, and survival.33 Decreased expressions of TLR2 have been reported in macrophages from cigarette smokers and patients with COPD.34 In addition, LPS stimulation on macrophages also does not increase TLR2 mRNA and protein expression. Alternatively, other studies have reported an upregulation of TLR2 in circulating blood neutrophils and peripheral blood-derived monocytes of patients with COPD.11,35 Similarly, Simpson et al,36 also reported an increased expression of TLR2 in COPD, which increases in severe to very severe stages. In accordance with the above-mentioned studies, our results also demonstrate increased expression of TLR2 in peripheral blood neutrophils of smokers with COPD.11,35,36 It is also increasing parallel and proportional to the disease severity. However, the difference was statistically insignificant. This may be due to the severity induced upregulation of TLR2 receptors in a small number of subjects in this group. These findings indicate that upon activation, neutrophils generate an innate immune response in an enhanced manner that is important in COPD pathogenesis.
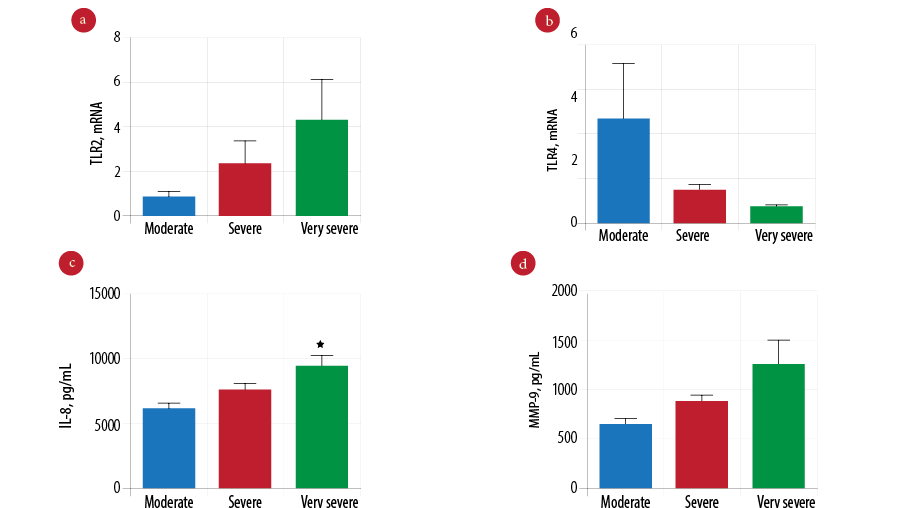
Figure 3: Comparison of innate immune mediators according to the severity of chronic obstructive pulmonary disease (COPD). Gene expression of Toll-like receptor 2 (TLR2) (a) and Toll-like receptor 4 (TLR4) (b) measured by real-time polymerase chain reaction (PCR) in moderate (n = 15), severe (n = 65), and very severe COPD patients (n = 21). Protein level of interleukin-8 (IL-8) (c) and mettaloproteinase-9 (MMP-9) (d) measured by enzyme-linked immunosorbent assay (ELISA) in moderate (n = 15), severe (n = 65), and very severe COPD patients (n = 21). Data presented as mean± standard error of the mean (SEM) and compared by one-way analysis of variance (ANOVA) test followed by Bonferroni post-test for multiple comparisons. *p-values < 0.050 are considered significant.
The role of TLR4 has been linked to COPD exacerbations in the context of bacterial infections and inflammations. Previous studies conducted on neutrophils and monocyte-derived macrophages demonstrating the involvement of TLR4 have reported the upregulated expression upon LPS stimulation in COPD.11,37 Similarly, another study reported a correlation between cigarette smoke exposure and the increased gene expression of TLR4 and TLR9 in addition to excess production of cytokine.38 Downregulation of TLR4 gene expressions was reported in the nasal epithelium of smokers and severe COPD patients compared to nonsmoking controls and patients with less severe COPD.39 We found the gene expression of TLR4 is higher in smokers with COPD compared to nonsmoking healthy controls. Interestingly, we found that the TLR4 expression dips during very severe COPD compared to moderate COPD; though it was still higher than nonsmoker controls. Although the response mechanism of innate immunity to cigarette smoke is unclear, it has been projected that injured airways epithelium produces a danger signal or DAMPs that may act as ligands for TLRs such as TLR2 and TLR4.40 This activates the transcription of proinflammatory cytokines such as IL-1, IL-6, and IL-8.39 We also observe the number of pack years smoked is positively correlated with inflammatory mediator IL-8 and negatively correlated with the severity of airflow obstruction. These results indicate that cigarette smoking may trigger the release of chemoattractant from epithelial cells. These chemoattractant may promote the neutrophils recruitment, which is related to the production of innate immune mediators that results in the decline of pulmonary functions.
Conclusion
This study demonstrates that LPS stimulation in peripheral blood neutrophil increases innate immune response, showing upregulation of TLR2 and TLR4 mRNA with increased release of IL-8 and MMP-9 protein in COPD. This outcome supports the possibility of activation of innate immune response, which is an important mechanism of disease pathogenesis. Henceforth, TLR2 and TLR4 may be considered as a diagnostic marker in COPD patients. Thus, we believe that TLR2 and TLR4 may be exploited in the future for therapeutic target discovery.
Disclosure
The authors declared no conflicts of interest. This study was funded by Indian Council of Medical Research (ICMR) New Delhi, India.
Acknowledgements
The authors acknowledge the staff in Department of Biochemistry for excellent technical assistance.
references
- 1. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Updated 2013 [cited 2013 March 3]. Available from http://www.csrd.org.cn/uploadfiles/img/file/20130305/20130305111459575957.pdf .
- 2. Sarir H, Henricks PA, van Houwelingen AH, Nijkamp FP, Folkerts G. Cells, mediators and Toll-like receptors in COPD. Eur J Pharmacol 2008 May;585(2-3):346-353.
- 3. Agusti A, Soriano JB. COPD as a systemic disease. COPD 2008 Apr;5(2):133-138.
- 4. Barnes PJ. New molecular targets for the treatment of neutrophilic diseases. J Allergy Clin Immunol 2007 May;119(5):1055-1062, quiz 1063-1064.
- 5. Larsson K. Aspects on pathophysiological mechanisms in COPD. J Intern Med 2007 Sep;262(3):311-340.
- 6. Texereau J, Chiche JD, Taylor W, Choukroun G, Comba B, Mira JP. The importance of Toll-like receptor 2 polymorphisms in severe infections. Clin Infect Dis 2005 Nov;41(Suppl 7):S408-S415.
- 7. Raoust E, Balloy V, Garcia-Verdugo I, Touqui L, Ramphal R, Chignard M. Pseudomonas aeruginosa LPS or flagellin are sufficient to activate TLR-dependent signaling in murine alveolar macrophages and airway epithelial cells. PLoS One 2009 Oct;4(10):e7259.
- 8. Wieland CW, Florquin S, Maris NA, Hoebe K, Beutler B, Takeda K, et al. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable haemophilus influenzae from the mouse lung. J Immunol 2005 Nov;175(9):6042-6049.
- 9. Reed CE, Milton DK. Endotoxin-stimulated innate immunity: A contributing factor for asthma. J Allergy Clin Immunol 2001 Aug;108(2):157-166.
- 10. Sabroe I, Lloyd CM, Whyte MK, Dower SK, Williams TJ, Pease JE. Chemokines, innate and adaptive immunity, and respiratory disease. Eur Respir J 2002 Feb;19(2):350-355.
- 11. Baines KJ, Simpson JL, Gibson PG. Innate immune responses are increased in chronic obstructive pulmonary disease. PLoS One 2011;6(3):e18426.
- 12. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med 2008 Jun;14(6):633-640.
- 13. Hansel TT, Barnes PJ .New drugs for exacerbations of chronic obstructive pumonary disease. Lancet 2009 Aug 29;374(9691):744-755.
- 14. O’Neill LA. The interleukin-1 receptor/toll-like receptor superfamily: 10 years of progress. Immunol Rev 2008 Dec;226:10-18.
- 15. Taggart CC, Greene CM, Carroll TP, O’Neill SJ, McElvaney NG. Elastolytic proteases: inflammation resolution and dysregulation in chronic infective lung disease. Am J Respir Crit Care Med 2005 May;171(10):1070-1076.
- 16. Owen CA. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2008;3(2):253-268.
- 17. Yamamoto C, Yoneda T, Yoshikawa M, Fu A, Tokuyama T, Tsukaguchi K, et al. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest 1997 Aug;112(2):505-510.
- 18. Oudijk EJ, Nijhuis EH, Zwank MD, van de Graaf EA, Mager HJ, Coffer PJ, et al. Systemic inflammation in COPD visualised by gene profiling in peripheral blood neutrophils. Thorax 2005 Jul;60(7):538-544.
- 19. Noguera A, Batle S, Miralles C, Iglesias J, Busquets X, MacNee W, et al. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001 Jun;56(6):432-437.
- 20. Clark RA, Nauseef WM. Isolation and functional analysis of neutrophils. Curr Protoc Immunol 2001 May;Chapter 7:Unit 7.23.
- 21. Reynolds PR, Cosio MG, Hoidal JR. Cigarette smoke-induced Egr-1 upregulates proinflammatory cytokines in pulmonary epithelial cells. Am J Respir Cell Mol Biol 2006 Sep;35(3):314-319.
- 22. Xu X, Wang H, Wang Z, Xiao W. Plasminogen activator inhibitor-1 promotes inflammatory process induced by cigarette smoke extraction or lipopolysaccharides in alveolar epithelial cells. Exp Lung Res 2009 Nov;35(9):795-805.
- 23. Noguera A, Busquets X, Sauleda J, Villaverde JM, MacNee W, Agustí AG. Expression of adhesion molecules and G proteins in circulating neutrophils in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998 Nov;158(5 Pt 1):1664-1668.
- 24. Noguera A, Sala E, Pons AR, Iglesias J, MacNee W, Agustí AG. Expression of adhesion molecules during apoptosis of circulating neutrophils in COPD. Chest 2004 May;125(5):1837-1842.
- 25. Petersen F, Van Damme J, Flad HD, Brandt E. Neutrophil-activating polypeptides IL-8 and NAP-2 induce identical signal transduction pathways in the regulation of lysosomal enzyme release. Lymphokine Cytokine Res 1991 Apr;10(1-2):35-41.
- 26. Garcia-Rio F, Miravitlles M, Soriano JB, Muñoz L, Duran-Tauleria E, Sánchez G, et al; EPI-SCAN Steering Committee. Systemic inflammation in chronic obstructive pulmonary disease: a population-based study. Respir Res 2010 May;11:63.
- 27. El-Shimy WS, El-Dib AS, Nagy HM, Sabry W. A study of IL-6, IL-8, and TNF-a as inflammatory markers in COPD patients. Egypt J Bronchol 2014 Aug;8(2):91-99.
- 28. Woessner JF Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J 1991 May;5(8):2145-2154.
- 29. Shapiro SD. Matrix metalloproteinase degradation of extracellular matrix: biological consequences. Curr Opin Cell Biol 1998 Oct;10(5):602-608.
- 30. Esa SA, Rawy AM, EL-Behissy MM, Kamel MH, El-Hwaitty HM. Study of the level of sputum matrix metalloproteinase-9 (MMP-9) and tissue inhibitor metalloproteinase-1 (TIMP-1) in COPD patients. Egyptian Journal of Chest Diseases and Tuberculosis 2014 Jul;63(4):861-867.
- 31. Eman Sobh, Almadbouly AA, Ezzat H, Abd-Allah M. Serum levels of high mobility group box 1 (HMGB1) and matrix metalloprotinase 9 (MMP9) are related to lung function indices in chronic obstructive pulmonary disease. Clinical Medicine and Diagnostics 2017;7(2):31-39.
- 32. Bezemer GF, Sagar S, van Bergenhenegouwen J, Georgiou NA, Garssen J, Kraneveld AD, et al. Dual role of Toll-like receptors in asthma and chronic obstructive pulmonary disease. Pharmacol Rev 2012 Apr;64(2):337-358.
- 33. Sabroe I, Jones EC, Whyte MK, Dower SK. Regulation of human neutrophil chemokine receptor expression and function by activation of Toll-like receptors 2 and 4. Immunology 2005 May;115(1):90-98.
- 34. Droemann D, Goldmann T, Tiedje T, Zabel P, Dalhoff K, Schaaf B. Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir Res 2005 Jul;6:68.
- 35. Pons J, Sauleda J, Regueiro V, Santos C, López M, Ferrer J, et al. Expression of Toll-like receptor 2 is up-regulated in monocytes from patients with chronic obstructive pulmonary disease. Respir Res 2006 Apr;7:64.
- 36. Simpson JL, McDonald VM, Baines KJ, Oreo KM, Wang F, Hansbro PM, et al. Influence of age, past smoking, and disease severity on TLR2, neutrophilic inflammation, and MMP-9 levels in COPD. Mediators Inflamm 2013 Mar;2013:462934.
- 37. Karimi K, Sarir H, Mortaz E, Smit JJ, Hosseini H, De Kimpe SJ, et al. Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir Res 2006 Apr;7:66.
- 38. Zuo L, Lucas K, Fortuna CA, Chuang C-C, Best TM. Molecular Regulation of Toll-like Receptors in Asthma and COPD. Front Physiol 2015 Nov;6:312.
- 39. MacRedmond RE, Greene CM, Dorscheid DR, McElvaney NG, O’Neill SJ. Epithelial expression of TLR4 is modulated in COPD and by steroids, salmeterol and cigarette smoke. Respir Res 2007 Nov;8:84.
- 40. Matzinger P. The danger model: a renewed sense of self. Science 2002 Apr;296(5566):301-305.