Feminizing adrenocortical carcinomas are defined as malignant tumors with estrogens overproduction with or without other adrenocortical hormones. They are rare even in endocrinology as they account for less than 1−2% of all adrenal tumors.1 They are mainly observed in men (median age = 42 years, range = 19−77 years).2 They are rare in children and exceptional in women. The prognosis is poor, especially when metastases are present
at diagnosis.
In males, the main symptom is gynecomastia, which may or may not be associated with other hypogonadism features. Our aim was to describe a case without gynecomastia and try to understand why this important clinical symptom was absent in our patient.
Case Report
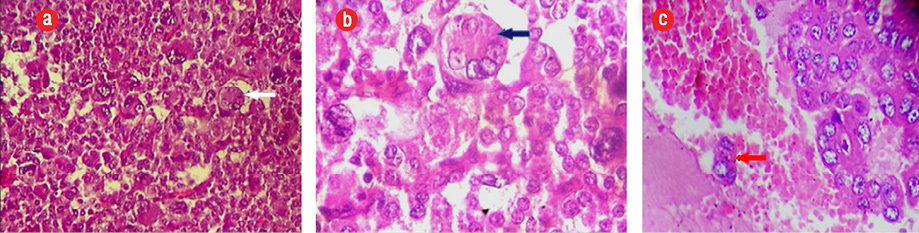
Figure 1: (a and b) Histological examination (hematoxylin and eosin staining) showing a malignant endocrine tumor with atypical cells, large nuclei with numerous mitotic figures (blue arrows), magnification = 10 ×, and (c) intestinal wall invasiveness (red arrow), magnification = 40 ×.
Table 1: Hormonal evaluation of the patient.
|
Estradiol |
645.32* |
55−165 pmol/L |
|
Cortisol level |
|
50−550 nmol/L |
|
8 AM
Midnight |
784.34
453.00 |
|
|
17OH Progesterone |
11.7 |
0.0−1.6 nmol/L |
|
Androstenedione |
8.82 |
0.3−3.1 ng/mL |
|
DHEA-S |
1337 |
95−530 µg/dL |
DHEA-S: dehydroepiandrosterone sulfate; *mean value.
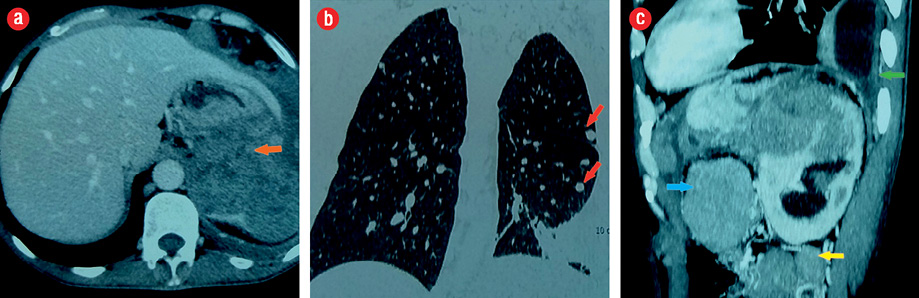
Figure 2: (a) A large adrenal tumor (orange arrow), (b) pulmonary metastases (red arrows), and (c) abdominal (blue arrow), retroperitoneal lymph nodes (yellow arrow), and pleural effusion (green arrow).
A 44-year-old man presented with abdominal pain. Radiological assessment revealed a large tumor measuring 120 × 95 mm in the left retroperitoneal area with numerous metastases. He was operated on without any hormonal assessment. Pathological examination showed an adrenal origin with a Weiss’s score of 5 [Figure 1]. Six months later, he had another surgery for a relapsing tumor and was sent to our department for hormonal evaluation. Clinical examination showed a skinny man with severe fatigue and anorexia. His body mass index (BMI) was 16 kg/m2, and his blood pressure was 110/60 mm. He had no Cushingoid features. Gynecomastia and galactorrhea were also absent. Penile length, testicular volume, and body hair growth were normal. However, many cutaneous nodules were present in the thoracic and abdominal areas. Hormonal assessment showed mixed adrenal secretion where estrogens were prevailing. He had high morning and midnight plasma cortisol, which failed to be suppressed with 2 mg dexamethasone, and very high estradiol and 17OH progesterone contrasting with low testosterone [Table 1]. Imaging studies revealed a relapsing tumor with numerous pleural, pulmonary, abdominal, and retroperitoneal metastases [Figure 2].
As Mitotane treatment was not available, he had chemotherapy (doxorubicin, carboplatin, and etoposide). He did not respond well to the treatment and died four months after diagnosis.
Discussion
Secreting and non-secreting adrenocortical carcinomas are rare malignant tumors with a dire prognosis,3,4 and typically occur in males in the fourth and fifth decades of life.3 Adrenocortical neoplasms may lead to a variety of endocrine syndromes depending on the type of produced hormones. When estrogens overproduction occurs, the term feminizing adrenal tumors (FATs) is used.5 Most of these tumors are malignant. Benign tumors, although rare, exist.
Feminizing adrenocortical carcinomas are exceedingly rare in adults and account for 1−2% of adrenocortical carcinomas.1 They are prevailing in adult males, although they can be observed in children. Female cases are exceptional.6,7
Patients with FATs usually complain of decreased libido and erection or ejaculation problems. They also have gynecomastia with or without other features of hypogonadism. In 1965, the first literature review recorded 52 FATs and demonstrated that patients with FATs have gynecomastia (98%), a palpable abdominal mass (58%), and a testicular atrophy (52%).8 Patients with FATs usually complain of diminished libido (48%) and breast tenderness (42%).8
On biological assessments, estrogens overproduction, with or without an increase in other adrenal hormones, are the main abnormalities. The increase in estrogens production results from peripheral androgens conversion, but also from direct production of the adrenocortical tumor.9 Gonadotropin hormones are normal-low or decreased. The response to luteinizing hormone-releasing hormone is generally blunted.
The main characteristic of these adrenocortical tumors is their ability to produce estrogens, although cortisol may be subnormal or high with or without clinical expression. Women can develop androgen excess. Estrogens overproduction occurs through an increase in androgen substrates such as androstenedione. However, it can also result from augmentation in aromatase activity within the tumor leading to an increased synthesis of estrone.1
Bilateral breast enlargement results from the imbalance between high estrogen and low free androgens. Gynecomastia is the major clinical expression.1 However, its absence does not eliminate the diagnosis of FATs. When gynecomastia is missing, one should discuss the resistance of breast tissue to estrogens, very rapid evolution, and severe protein degradation.
FATs have a very poor prognosis. Mortality among adult males with FATs is high as the three-year survival is less than 20% after tumor resection7 especially in patients diagnosed late with multiple abdominal or extra-abdominal metastases.
Common sites of intra-abdominal metastases include the liver, intestine, peritoneum, and retroperitoneal lymph nodes. Extra-abdominal metastases may be observed in the lungs, pleura, bones, and other sites.
Treatment is based mainly on surgery with complete resection of the tumor and its metastases when possible. For recurrent or metastatic FATs, radiation therapy and various chemotherapies can be considered. However, with the exception of Mitotane, there is no other effective medication.5 Aromatase inhibitors have been used
without success.5
Conclusion
Feminizing adrenal carcinomas are exceptional and have a dire prognosis. Gynecomastia, which is the classical manifestation, was absent in this patient probably due to the resistance of the breast tissue
to estradiol.
Disclosure
The authors declared no conflicts of interest.
Acknowledgements
The authors would like to thank Prof. Kheira Bendissari, head of the department of pathology and Prof. Mustapha Boubrit, head of the department of radiology for providing pathological and radiological images, respectively.
references
- Moreno S, Guillermo M, Decoulx M, Dewailly D, Bresson R, Proye Ch. Feminizing adreno-cortical carcinomas in male adults. A dire prognosis. Three cases in a series of 801 adrenalectomies and review of the literature. Ann Endocrinol (Paris) 2006 Mar;67(1):32-38.
- 2. Chentli F, Bekkaye I, Azzoug S. Feminizing adrenocortical tumors: Literature review. Indian J Endocrinol Metab 2015 May-Jun;19(3):332-339.
- 3. Gogoi G, Baruah MP, Borah P, Borgohain M. Adrenocortical carcinoma: An extremely uncommon entity and the role of Immunohistochemistry in its diagnosis. Indian J Endocrinol Metab 2012 Dec;16(Suppl 2):S373-S375.
- 4. Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab 2006 Jun;91(6):2027-2037.
- 5. Kidd MT, Karlin NJ, Cook CB. Feminizing adrenal neoplasms: case presentations and review of the literature. J Clin Oncol 2011 Feb;29(6):e127-e130.
- 6. Barcelo B, Abascal J, Ardaiz J, Gil P, Colas A, Menendez J, et al. Feminizing adrenocortical carcinoma in a postmenopausal woman. Postgrad Med J 1979 Jun; 55(644):406-408.
- 7. Bhettay E, Bonnici F. Pure oestrogen-secreting feminizing adrenocortical adenoma. Arch Dis Child 1977 Mar;52(3):241-243.
- 8. Gabrilove JL, Sharma DC, Wotiz HH, Dorfman RI. Feminizing adrenocortical tumors in the male: A review of 52 cases including a case report. Medicine (Baltimore) 1965 Jan;44:37-79.
- 9. Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994 Apr;17(4):275-278.