Primary hepatic lymphoma (PHL) is a tumor confined to the liver without evidence of lymphomatous involvement of the spleen, lymph nodes, bone marrow, or other lymphoid structures.1 PHL is a rare type of extranodal lymphoma accounting for less than 1% of all lymphomas.2 It presents with a wide range of subtle manifestations and, for this reason, the diagnosis may be delayed or missed. Diagnosis is confirmed by liver biopsy, and a review of the literature showed success with certain surgical and chemotherapeutic treatments. Most patients are treated with chemotherapy.
The majority of PHL cases originate from B-cells, while T-cell lymphoma is less common.3 Misdiagnosis is frequent, and cases of unnecessary resection have been reported.2 The purpose of this case report is to describe a case of PHL and emphasize the importance of accurate diagnosis before implementing a therapeutic plan.2
Case report
A 67-year-old male presented with a three-week history of jaundice, pain in the epigastric and right upper quadrant regions, nausea, and vomiting. His symptoms did not improve with omeprazole treatment, and generalized pruritus and icterus developed. His medication history was negative. He did not smoke or use illicit drugs. No previous illnesses were reported. His familial history for malignancy was negative.
On examination, he was febrile (38.1 °C), and his abdomen was soft with tenderness in the right upper quadrant and epigastric region. His spleen was not palpable. There were excoriation scars due to itching on the limbs. Other examinations were normal. Blood tests on admission showed total bilirubin levels of 19.1 mg/dL and direct bilirubin level of 11.7 mg/dL, aspartate aminotransferase of 48 U/L, alanine aminotransferase of 36 U/L, and alkaline phosphatase (ALP) of 535 U/L. His white blood cell count was 11 300/mL, hemoglobin was 10.4 g/dL, and platelets were 217 000/mL. His amylase level was 59 mg/dL, and lactate dehydrogenase (LDH) was 515 U/L. Ultrasonography of abdomen revealed dilatation of the intra- and extra-hepatic bile ducts and a gallbladder with a normal intramural diameter. There was one hypoechoic oval shape lesion with sharp borders (11 × 19 mm) around the hepatic hilum and one (15 mm) impacted stone in the neck of the gallbladder.
Computed tomography (CT) scans of the abdomen and pelvis with contrast (intravenous and oral) showed a normal sized liver and a large mass lesion with infiltrative appearance and ill-defined margins in the central portion of the liver, extending to the right and left liver lobe, which showed mild enhancement with the contrast. The image was suggestive of cholangiocarcinoma (Klatskin tumor) [Figure 1]. The right and left hepatic ducts and proximal of the common hepatic duct were involved in the mass, resulting in dilatation of intrahepatic ducts. The common bile duct was normal. The gallbladder had a thick, edematous wall and fluid was seen in gallbladder fossa. There were a few small stones in gallbladder suggestive of acute cholecystitis. No lymphadenopathy was seen.
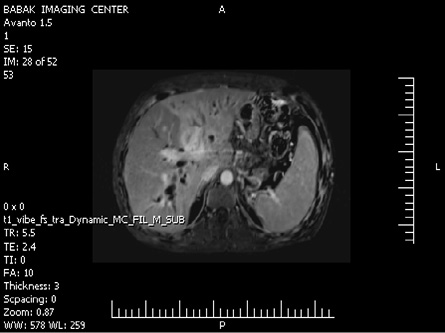
Figure 1: Contrast-enhanced computed tomography of the abdomen showing the hepatic lesion.
An endoscopic retrograde cholangiopancreatography was performed. A biliary stent was inserted, and a specimen for cytological test was collected by brushing the biliary ducts. Cytology was negative for malignancy. Serology for hepatitis A, B, and C were negative. Levels of alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA), and carbohydrate antigen 19-9 (CA 19-9) were normal. The patient was treated with antibiotics (ceftriaxone and metronidazole) for seven days, and a cholecystectomy and liver mass biopsy were performed. Pathological report of the gallbladder and wedge biopsy of liver hilar mass after immunohistochemistry showed large B-cell lymphoma (positive for CD20+ lymphocytes) and acute chronic cholecystitis [Figure 2, 3, and 4]. His bone marrow biopsy was normal. He was started on combination chemotherapy of rituximab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunomycin), vincristine sulfate (Oncovin), and prednisone (R-CHOP). The patient showed a good response to the treatment with mild side effects.
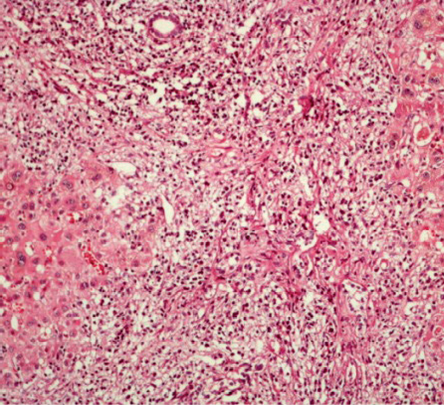
Figure 2: Liver biopsy cytology showing lymphoma cells with hyperchromatic nuclei and little cytoplasm stained with hematoxylin and eosin, magnification = 40 ×.
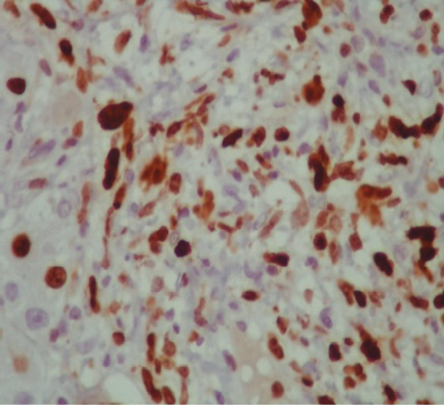
Figure 3: Liver biopsy cytology with positive staining for Ki 673 (a cell proliferation marker present in the nuclei of cells in the mitosis), magnification = 40 ×.
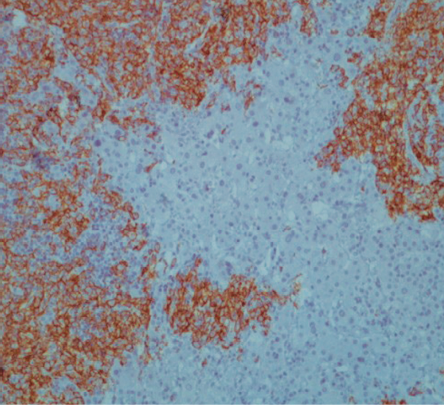
Figure 4: Liver biopsy immunostaining for CD20 (B-lymphocyte marker). Strong membrane and cytoplasmic positivity confirmed B-cell lymphoid proliferation, magnification = 20 ×.
Discussion
PHL is different from other non-PHLs, which involves the liver in 50% of cases.3 To make a diagnosis, there must be no nodal involvement, involve predominantly the liver, and the absence of any evidence of leukemia. Clinical features and laboratory tests are wide ranging, and liver biopsy is essential to confirm a suspected diagnosis.
Although rare, it is important to recognize PHL because it responds favorably to chemotherapy and may have a better prognosis than hepatocellular carcinoma or metastatic disease of the liver. The differential diagnosis should include primary hepatoma and metastatic neoplasms. When imaging findings on CT scans and magnetic resonance imaging (MRI) are nonspecific, a biopsy is needed not only for a definitive diagnosis but also for identifying the immunophenotype of the PHL. These type of lesions are highly chemosensitive, and early aggressive chemotherapy may result in sustained remission.1,4
PHL is more frequent in men, and the usual age of presentation is around 50 years. Presentations vary from the incidental discovery of hepatic abnormalities in asymptomatic patients to the onset of fulminant hepatic failure with rapid progression of encephalopathy to coma and death. Symptoms are usually nonspecific and include right upper quadrant and epigastric pain, fatigue, weight loss, fever, anorexia, and nausea. Hepatomegaly is very common, and jaundice may be found on physical examination.1,4
PHL can be subdivided into nodular or diffuse types according to the presence of liver infiltration. Most PHL corresponds to a larger cell type and demonstrates a B-cell immunophenotype. Other histologic subtypes of PHL include high-grade tumors (e.g., lymphoblastic and Burkitt lymphoma, 17%), follicular lymphoma (4%), diffuse histiocytic lymphoma (5%), lymphoma of the mucosa-associated lymphoid tissue, anaplastic large-cell lymphoma, mantle cell lymphoma, and T-cell/histiocytes-rich B-cell lymphoma.1,4 The diagnosis of PHL remains a challenge. Misdiagnosis is common and many times is made by histology of the surgical specimen. Because of the presence of a large area of necrosis, the fine needle biopsy is frequently negative. During the procedure, one should be careful to guide the needle toward an area without necrosis to get a representative sample of the tumor.
Patients with PHL typically have abnormal liver function tests, with elevated LDH and ALP. Elevated LDH with normal AFP and CEA remains a valuable biologic feature.1 On ultrasound, PHL lesions are hypoechoic compared to normal liver. Imaging by CT shows hypoattenuating lesions and MRI enhancement after contrast. Findings on MRI are variable; however, a few authors have described hypointense T1-weighted images and hyperintense T2-weighted images.1,4−6 For PHL diagnosis, the tumor must be confined to the liver, without involvement of the spleen, lymph nodes, bone marrow, or other lymphoid structures.5 The primary treatment in patients with PHL is chemotherapy. Other therapeutic modalities reported in the literature are surgical resection, if possible, radiotherapy, biliary drainage procedures for the treatment of jaundice with external-internal drain, biliary stent placement, and percutaneous drain placement.1,5−9
The standard treatment for patients with diffuse large B-cell lymphoma is combination chemotherapy (R-CHOP). Poor prognostic features include advanced age, constitutional symptoms, bulky disease, unfavorable histologic subtype, elevated levels of LDH and a high proliferation rate, cirrhosis, and comorbid conditions.1,5−7
Conclusion
PHL is rare but should be considered in differential diagnosis of patients presenting with biliary obstruction. Hepatic hilum is one of common locations of the biliary system that can be compressed by lymphoma, and the mainstay of treatment is chemotherapy.
Disclosure
The authors declared no conflicts of interest.
references
- Resende V, Oliveira TS, Gomes RT, Laboissière RS, Tavares-Junior WC, de Melo Couto OF. Primary hepatic lymphoma: A case report. Int J Surg Case Rep 2013;4(12):1165-1168.
- 2. Haider FS, Smith R, Khan S. Primary hepatic lymphoma presenting as fulminant hepatic failure with hyperferritinemia: A case report. J Med Case Reports 2008 Dec;2:279.
- 3. Delshad SD, Ahdoot JJ, Portocarrero DJ. Primary hepatic lymphoma. Clin Gastroenterol Hepatol 2010 May;8(5):e49-e50.
- 4. Masood A, Kairouz S, Hudhud KH, Hegazi AZ, Banu A, Gupta NC. Primary non-Hodgkin lymphoma of liver. Curr Oncol 2009 Aug;16(4):74-77.
- 5. Ma YJ, Chen EQ, Chen XB, Wang J, Tang H. Primary hepatic diffuse large B cell lymphoma: A case report: Primary hepatic diffuse large B cell lymphoma. Hepat Mon 2011 Mar;11(3):203-205.
- 6. Gomyo H, Kagami Y, Kato H, Kawase T, Ohshiro A, Oyama T, et al. Primary hepatic follicular lymphoma : a case report and discussion of chemotherapy and favorable outcomes. J Clin Exp Hematop 2007 Nov;47(2):73-77.
- 7. Doi H, Horiike N, Hiraoka A, Koizumi Y, Yamamoto Y, Hasebe A, et al. Primary hepatic marginal zone B cell lymphoma of mucosa-associated lymphoid tissue type: case report and review of the literature. Int J Hematol 2008 Nov;88(4):418-423.
- 8. Basir N, Bickle IC, Telisinghe PU, Abdullah MS, Chong VH. Signet Ring Cell Lymphoma of the Small Bowel: A case report. Oman Med J 2012 Nov;27(6):491-493.
- 9. Zentar A, Tarchouli M, Elkaoui H, Belhamidi MS, Ratbi MB, Bouchentouf SM, et al. Primary hepatic lymphoma. J Gastrointest Cancer 2014 Sep;45(3):380-382.