| |
Introduction
A 28-year-old female presented with recurrent epistaxis and mild hemoptysis for two years. There was no fever, dyspnea, palpitations, obstructed breathing or hematuria. There was no history of past tuberculosis or drug intake. Both her parents died when she was 5-years of age from suspected tuberculosis. On examination, she had pallor and nasal telangiectasia.
Blood pressure and pulse rate was normal. There was no clubbing, cyanosis or pedal edema. Cardiovascular system examination was normal. Laboratory investigations revealed normal hematological profile with a hemoglobin of 12.5 g, platelet count of 250,000/μl, and prothrombin time of 12 seconds. Chest radiograph showed ill-defined nodules in the right middle and left lower zones (Fig. 1). A contrast enhanced computed tomography (CECT) of the chest was undertaken. Sequential axial CECT sections (Figs. 2a and b) of the chest in lung and mediastinal window settings are shown below..
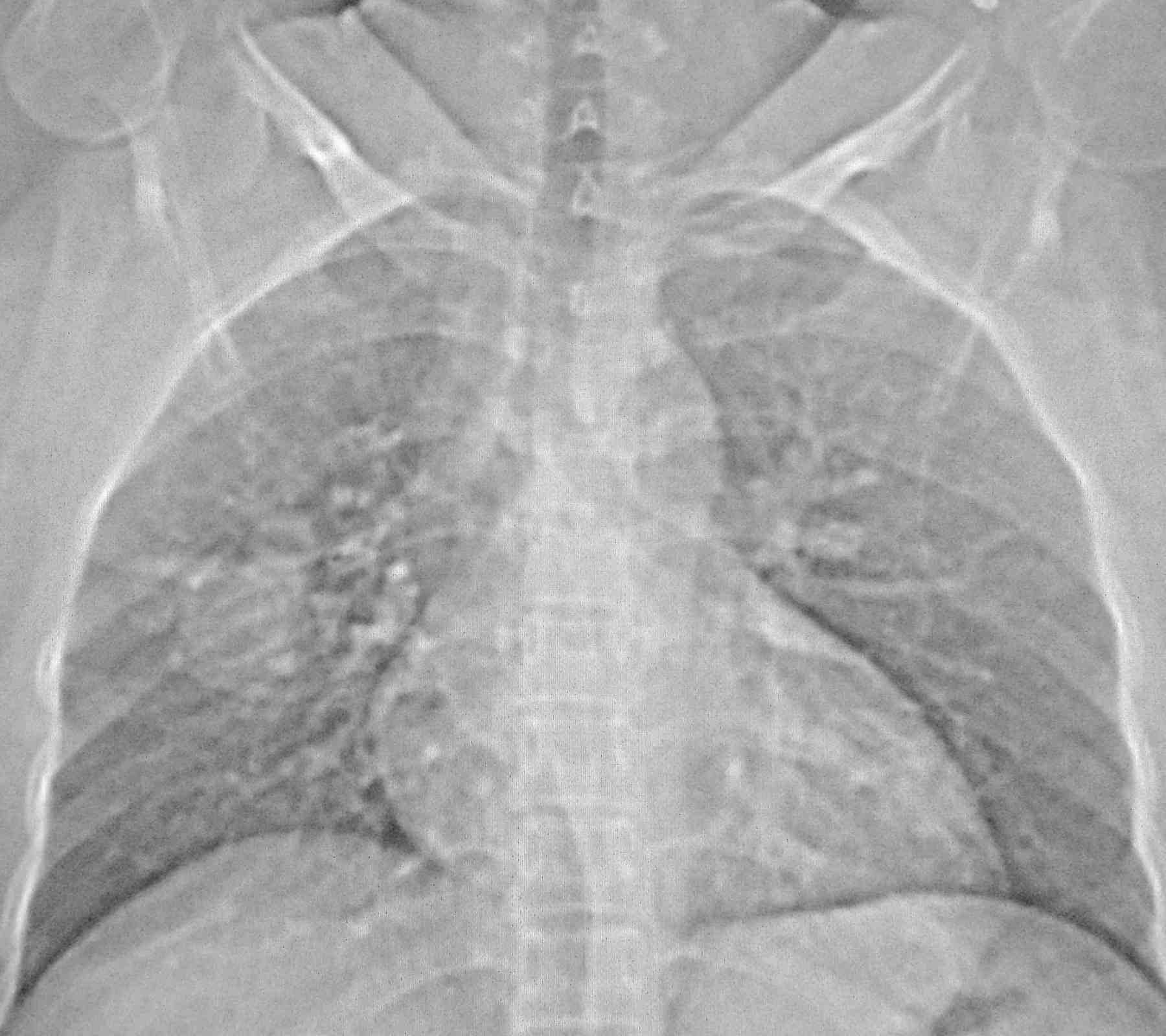
Figure 1: Chest Radiograph.
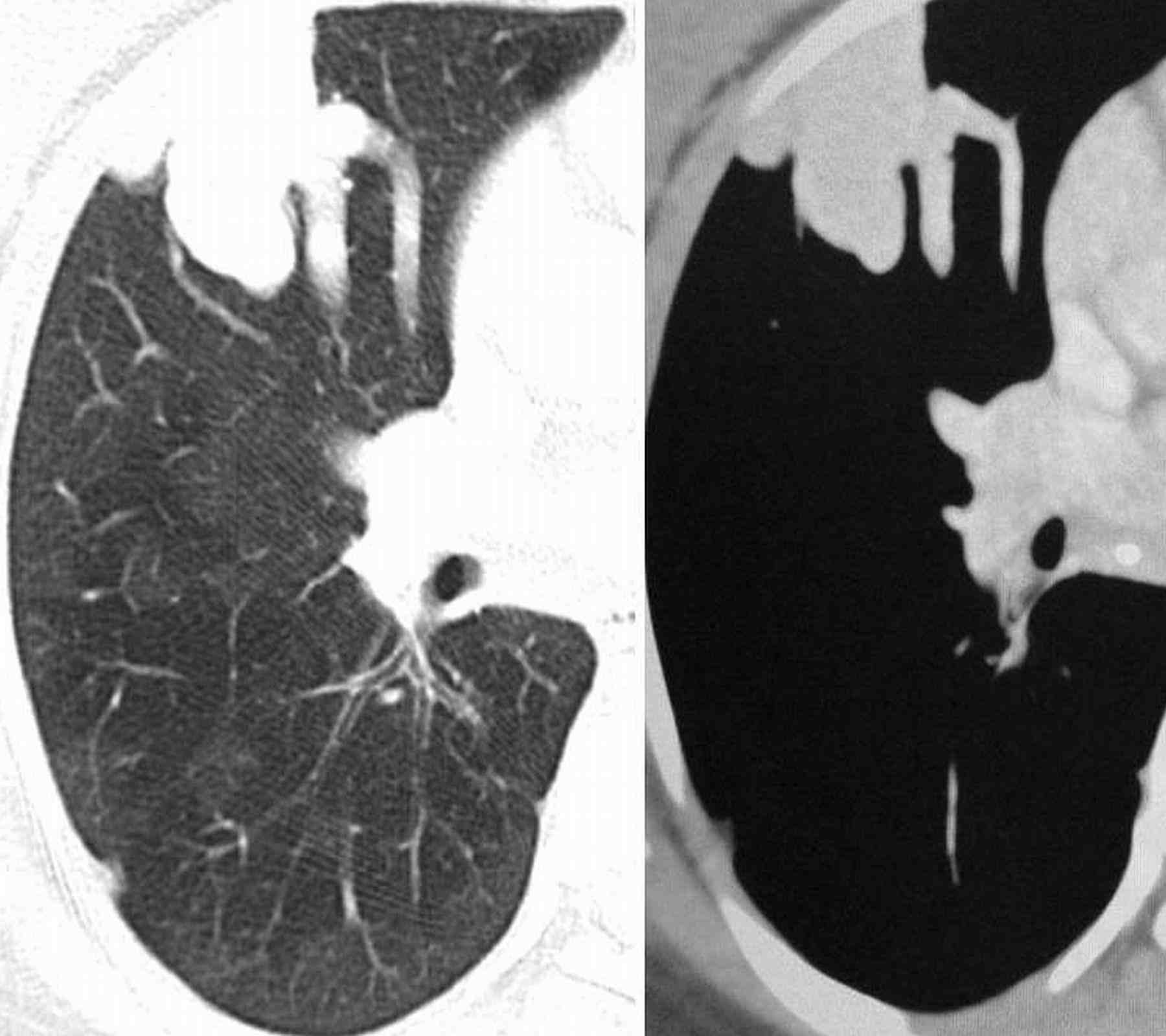
2a
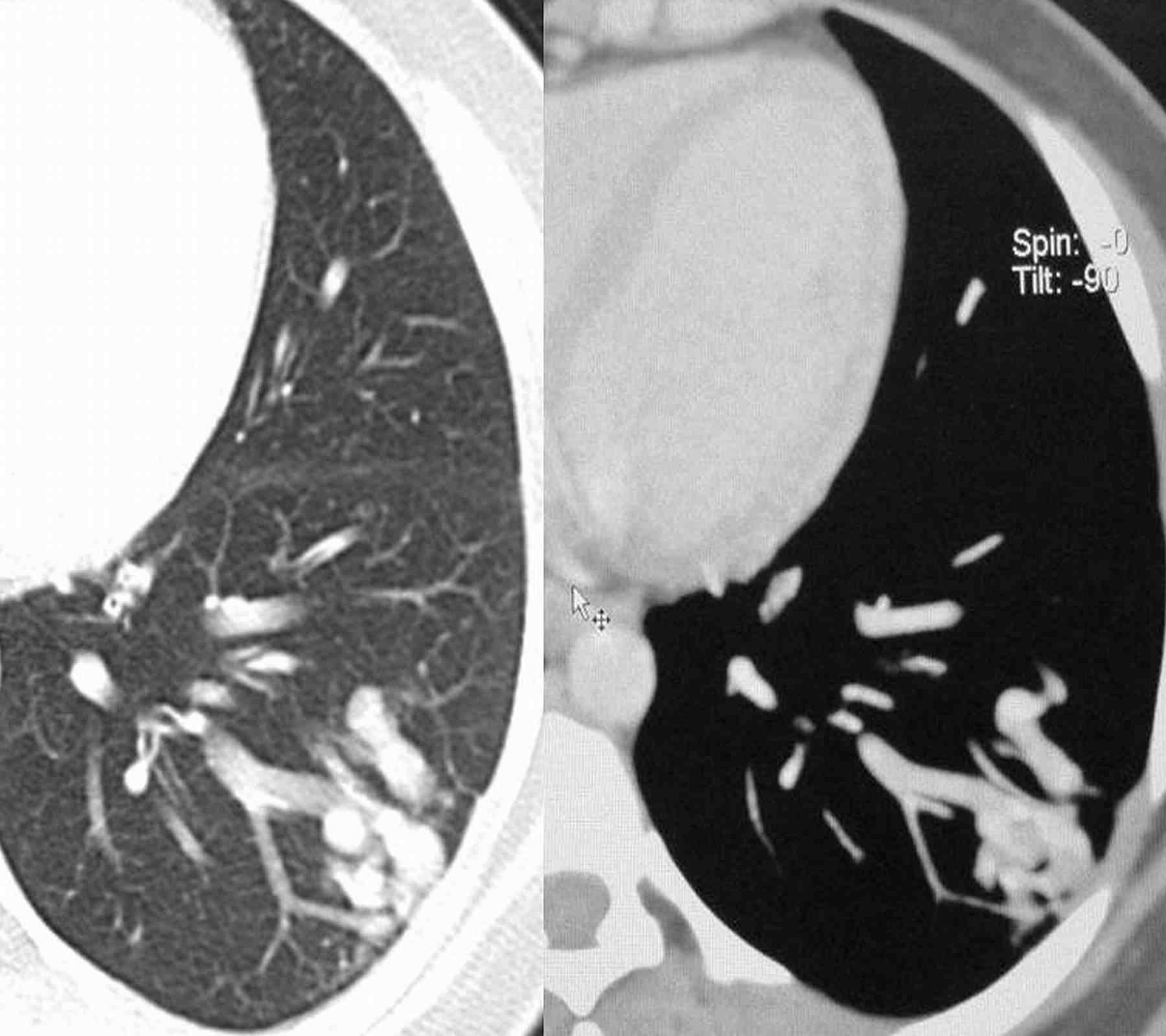
2b
Figure 2a & b: Sequential axial CECT sections.
Question
What is your diagnosis?
A. Wegener’s granulomatosis
B. Bronchiectasis
C. Multiple arterio-venous malformations
D. Metastases
E. Rheumatoid nodule
Answer
C. Multiple arterio-venous malformations
Discussion
Pulmonary arteriovenous malformations (PAVMs) are rare. Most are congenital associated with hereditary haemorrhagic telangiectasia (HHT). Acquired causes include: chest trauma, cardiovascular interventional procedures, chronic cirrhosis, mitral stenosis, and schistosomiasis. HHT is responsible for 80% of PAVMs and PAVMs are present in 15-45% of patients with HHT.1 HHT, or Rendu-Osler-Weber disease, is an autosomal dominant multiorgan fibrovascular dysplasia with a reported prevalence of 1 in 10,000 to 1 in 5000.2 The anatomic structure of the PAVMs in HHT is classified into two types,3 simple, with a single feeding artery with a single draining vein and complex, with two or more arterial branches, and two or more draining veins. Mutations associated with HHT include: endoglin mutation, activin receptor-like kinase (ALK)-1 mutation and SMAD4 mutation.4
Epistaxis secondary to nasal mucosa telangiectasia is the most common manifestation. Clinical manifestations related to pulmonary involvement include dyspnea, cyanosis, clubbing and hemoptysis. Complications associated with untreated PAVMs are life threatening hemorrhage and neurological complications. Neurological complications include transient ischaemic attack and stroke; results from the paradoxical embolization of leg vein thrombus to the left-sided arterial circulation through the PAVMs. Rarely, the thrombus can originate in situ in PAVMs. Another common neurological complication is cerebral abscess secondary to paradoxical embolization of bacterial biofilms.
Diagnosis is based on the Curaçao Criteria established by the Scientific Division of the Hereditary Hemorrhagic Telangiectasia International Foundation.5 Diagnosis is made in the presence of three of the following: recurrent spontaneous epistaxis, mucocutaneous telangiectasis, an autosomal dominant familial distribution, and visceral AVMs. Screening for PAVMs is recommended in all HHT patients due to the serious complications associated with them and the availability of safe and effective interventions.6 The screening method of choice is Contrast echocardiography with a sensitivity of 90%.1 Unenhanced Computed tomography (CT) thorax is used to confirm diagnosis and measure the size of PAVMs in patients with positive screening echocardiography. With the availability of multidetector CT (MDCT), contrast enhancement is not required for detection of PAVMs.7 On CT, the findings include a serpiginous mass or nodule with one or more enlarged feeding arteries and one or more draining veins (depending on whether the malformation is simple or complex). Embolization of PAVM with coils is the treatment of choice.2 A feeding artery calibre of at least 3 mm is considered the threshold for treatment of PAVMs.5 Recurrence of PAVMs occurs in around 19% of treated patients. Therefore, follow-up CTs are recommended at 1 year and every 3-5 years thereafter.
Other visceral manifestations include central nervous system manifestations: cerebral vascular malformations, spinal cord vascular malformation, encephalopathy, brain abscess and stroke.8 Liver is involved in 30% to 73% of patients with HHT. Hepatic fibrovascular dysplasia ranges from small telangiectases to large vascular masses.9 Recurrent gastrointestinal bleeding from mucosal fibrovascular dysplasia occurs in about 20% of patients with HHT.
Based on the patient’s presentation with epistaxis and hemoptysis; clinical differential diagnoses of pulmonary tuberculosis, Wegener’s granulomatosis, bleeding disorders, drug intake (drugs affecting the coagulation profile), and collagen vascular disorders were considered. Radiological differential diagnoses of multiple lung nodules on chest radiograph include neoplastic lesions (metastases, synchronous lung cancers), multiple pulmonary arterio-venous malformations, infections (multiple abscesses, fungal infections, hydatid disease) and immunological conditions (wegener’s granulomatosis, rheumatoid nodules, sarcoidosis, amyloidosis, organising pneumonia).10
Acknowledgements
The authors reported no conflict of interest and no funding was receive on this work.
References
1. Cottin V, Plauchu H, Bayle JY, Barthelet M, Revel D, Cordier JF. Pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. Am J Respir Crit Care Med 2004 May;169(9):994-1000.
2. Cottin V, Chinet T, Lavolé A, Corre R, Marchand E, Reynaud-Gaubert M, et al; Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia: a series of 126 patients. Medicine (Baltimore) 2007 Jan;86(1):1-17.
3. White RI Jr, Lynch-Nyhan A, Terry P, Buescher PC, Farmlett EJ, Charnas L, et al. Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radiology 1988 Dec;169(3):663-669.
4. Letteboer TG, Mager JJ, Snijder RJ, Koeleman BP, Lindhout D, Ploos van Amstel JK, et al. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J Med Genet 2006 Apr;43(4):371-377.
5. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000 Mar;91(1):66-67.
6. Gossage JR. Primary pulmonary hypertension or portopulmonary hypertension? Chest 1998 Oct;114(4):1224-1225.
7. Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI Jr. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radiol 2006 Jan;17(1):35-44, quiz 45.
8. Haitjema T, Westermann CJ, Overtoom TT, Timmer R, Disch F, Mauser H, et al. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease): new insights in pathogenesis, complications, and treatment. Arch Intern Med 1996 Apr;156(7):714-719.
9. Siddiki H, Doherty MG, Fletcher JG, Stanson AW, Vrtiska TJ, Hough DM, et al. Abdominal findings in hereditary hemorrhagic telangiectasia: pictorial essay on 2D and 3D findings with isotropic multiphase CT. Radiographics 2008 Jan-Feb;28(1):171-184.
10. Siddiki H, Doherty MG, Fletcher JG et al. Abdominal findings in hereditary hemorrhagic telangiectasia: pictorial essay on 2D and 3D findings with isotropic multiphase CT. Radiographics 2008; 28:171–184
|
|