
Sanjad-Sakati Syndrome in Omani children
doi:10.5001/omj.2010.63
ABSTRACT
Sanjad Sakati Syndrome is an Autosomal Recessive disorder found exclusively in people of Arabian origin. It was first reported from the Kingdom of Saudi Arabia in 1988. This is a report of a family with this rare disease in Oman. The syndrome comprises of congenital hypoparathyroidism, severe growth retardation, low IQ and typical facial features. Supportive treatment in the form of vitamin D and growth hormone is often offered to these children.
From the Department of Child Health, Sultan Qaboos University Hospital,
Al Khod, Muscat, Sultanate of Oman.
Received: 17 Jan 2010
Accepted: 30 Mar 2010
Address correspondence and reprint request to: Dr. Saif Al-Yaarubi, Department of Child Health, Sultan Qaboos University Hospital, Al Khod, Muscat, Sultanate of Oman.
E-mail: hawa34@hotmail.com
INTRODUCTION
Sanjad Sakati syndrome (SSS) is a newly described syndrome mainly from the Middle East and the Arabian Gulf countries.1-8 Children affected with this condition are born IUGR and present with hypocalcaemic tetany or seizures due to hypoparatyroidism at an early stage in their lives. They have typical physical features, namely; long narrow face, deep set small eyes, beaked nose, large floppy ears and micrognathia- severe failure to succeed and mild to moderate mental retardation.
CASES REPORT
Three Omani siblings (two girls and one boy) presented to the Pedicatrics department at Sultan Qaboos University Hospital with SSS. The children were thin and lean with narrow faces, deep set small eyes, bowed nasal bridges, pointed noses, large ears, long philtrum, thin lips, long tapering fingers, clinodactyly, microcephaly with small hands and feet. The boy had pinoscrotal hypoplasia and short penis which was later surgically corrected, while the girls had normal genitalia. All other systems were normal.
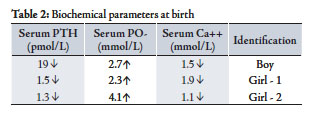
The boy had convulsions at the age of one month. Investigations showed: Low calcium - 1.5 mmol/L, High Phosphate - 2.7 mmol/L , normal Alkaline phosphatase and Low PTH - 19 pmol/L (n= 29-85). The convulsions were controlled with intravenous calcium infusions. Later, he required calcium and one alpha Alfacalcidol to maintain his calcium levels. Once his calcium was maintained, oral calcium was withdrawn. He had convulsions at the age of 3 months, 6 months and 4 years because of low calcium responded to IV calcium.
Girl - 1 developed convulsions on the third day of life. Her calcium profile showed: Calcium - 1.9 mmol/L, Phosphate - 2.3 mmol/L, normal Alkaline phosphatase and PTH 1.5 pmol/L. (n=1.3 - 5.4). She responded well to calcium infusion. She was also started on oral calcium and alphacalcidol. Later on calcium was stopped and she maintained her serum calcium on alphacalcidol only.
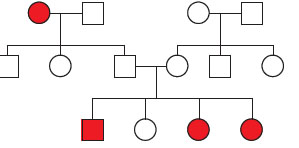
Figure 1: Family Pedigree
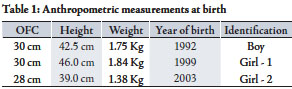
Table 1: Anthropometric measurements at birth
Girl – 2; She was investigated because of family history. She also showed the same biochemical picture: Ca - 1.1 mmol/L, PO4 - 4.1 mmol/L and PTH - 1.3pmol/L. Calcium and alphacalcidol was started, (Table 2). She had two episodes of hypocalcemic convulsions, requiring intravenous calcium. After the age of one year, she was able to maintain normal calcium and phosphate levels on one alphacalcidol only, like her brother and sister.
The siblings were extensively investigated for dysmorphology including extremely short stature. Endocrine work up showed normal thyroid functions but growth hormone results were borderline.
Ultrasound of the head and abdomen, chest X-ray, skeletal survey and karyotypes were normal. Other tests performed included flow cytometry for lymphocyte subsets, Immunoglobulin level and ECHO, and were found to be normal.
Table 2: Biochemical parameters at birth
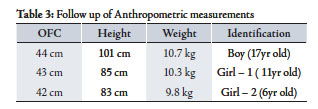
They all had low intelligence quotient of around 50 on the Stanford Binet Intelligent Scale. They had mild hypermetropia. The parents were counselled regarding the trial of growth hormone. Growth hormone was started for the boy for approximately one year at a dose of 0.03 mg/kg /day but the response to the treatment was minimal. On follow up, the growth of these patients was severely affected. (Table 3)
Table 3: Follow up of Anthropometric measurements
The boy was recently diagnosed with central hypoventilation on sleep polysomonography study and there were plans to start him on home ventilation. However, the boy recently presented with severe pneumonia requiring intensive care admission and ventilation. The child passed away during this admission at the age of 17 years due to respiratory compromise and multiorgan failure post pneumonia.
The patients were recognized due to their classic phenotypic presentation and the biochemical abnormalities. IUGR lead to severe growth retardation, facial dysmorphism and lifelong low IQ. Severe hypocalcaemia required treatment with an active form of vitamin D and calcium initially intravenously and then orally. Calcium was weaned off but they remained on vitamin D. Growth hormone trial was given for one year but there was minimal response.
DISCUSSION
Sanjad-Sakati Syndrome is an Autosomal Recessive disorder reported exclusively from the Arabian Peninsula. Reported patients have been Saudi, Qatari, Israeli Arabs, Kuwaiti and now Omani.1-8
The condition was first reported by Sanjad et al. in 1988.1 Three years later, its inheritance and configuration was confirmed by the same team from King Faisal Specialist Hospital and Research Centre, Saudi Arabia.2 Later on, the syndrome was also described by Richardson and Kirk in 1990, 3 and in 1992 by Kalam and Hafeez.4
SSS is listed in OMIM (241410) as HRD, that is, Hypoparathyroidism-Retardation-Dysmorphism Syndrome. Parvari et al. reported a gene location on chromosome number 1 at 1q42-43 and caused by mutations in the TBCE gene.5
SSS consists of hypoparathyroidism leading to hypocalcaemic tetany, seizures or both, intrauterine growth retardation which continues throughout the whole life. Patients have typical facial features and are mentally subnormal. The patients usually have characteristic features such as long narrow face, deep set eyes, beaked nose, large floppy ears, long filtrum, thin upper lip and micrognathia. Usually they are symptomatic in the new born period with complications of hypocalcaemia and investigations reveal a picture of hypoparathyroidism.
Although some of the features resemble DiGeorge Syndrome, Kenny-Caffey Syndrome and familial Hypoparathyroidism, no association with cardiac lesion, lymphopenia or skeletal abnormalities makes it a distinct entity.
Marsden et al. reported the use of growth hormone for a child with SSS with good outcome.6 The first child in this family was treated with growth hormone but the there was a little benefit in height. Although central hypoventilation has not been frequently reported in the literature for this syndrome, patients need to be investigated early in life and home ventilation should be provided to prevent further complications.
CONCLUSION
This is the first report of Sanjad Sakati syndrome in the Sultanate of Oman. In the future there are plans to arrange for DNA analysis to confirm the disease and to provide proper counselling of the families. Early recognition of the disease will lead to proper treatment of patients and prevent associated comorbidities.
ACKNOWLEDGEMENTS
The authors reported no conflict of interest and no funding was received on this work.
-
Sanjad S, Sakati N, Abu-Osba Y. Congenital hypoparathyroidism with dysmorphic features: a new syndrome. (Abstract) Pediat. Res 1988; 23: 271A.
-
Sanjad SA, Sakati NA, Abu-Osba YK, Kaddora R, Milner RDG. A new syndrome of congenital hypoparathyroidism, seizure, growth failure and dysmorphic features. Arch. Dis. Child 1991; 66: 193-196.
-
Richardson RJ, Kirk JMW. A new short stature, mental retardation syndrome, with hypoparathyroidism. Archives of Diseases in Childhood, 1990; 65: 1113-1117.
-
Kalam MA, Hafeez W. Congenital hypoparathyroidism, seizure, extreme growth failure with developmental delay and dysmorphic features-another case of this new syndrome. Clin Genet 1992; 42:110-113.
-
Parvari, R.; Hershkovitz, E.; Kanis, A.;Gorodischer, R.; Shalitin, S.; Sheffield, V. C.; Carmi, R. :Homozygosity and linkage-disequilibrium mapping of the syndrome of congenital hypoparathyroidism, growth and mental retardation, and dysmorphism to a 1-cM interval on chromosome on chromosome 1q42-43.Am. J. Hum. Genet. 63: 163-169, 1998.
-
Kelly TE, Blanton S, Saif R, Sanjad SA, Sakati N. Confirmation of the assignment of the Sanjad-Sakati (congenital hypoparathyroidism) syndrome (OMIM 241410) locus to chromosome lq42-43. Journal of Medical Genetics 2000; 37:63-64.
-
Marsden D, Nyhan WL, Sakati NO. Syndrome of hypoparathyroidism, growth hormone deficiency, and multiple minor anomalies. Am. J. Med. Genet. 1994; 52:334-338.
-
Naguib KK, Gouda SA, Elshafey A, Mohammed F, Bastaki L, Azab AS, et al. Sanjad–Sakati syndrome/Kenny–Caffey syndrome type 1: a study of 21 cases in Kuwait. East Mediterr Health J 2009 Mar; 15(2):345-352.
How to cite this article
Rafique B, Al-Yaarubi S. Sanjad-Sakati Syndrome in Omani children. OMJ 2010 July; 25(3):227-229.
How to cite this URL
Rafique B, Al-Yaarubi S. Sanjad-Sakati Syndrome in Omani children. OMJ [online] 2010 July; 25(3):227-229. Available at http://www.omjournal.org/CaseReports/FullText/201007/FT_SanjadSakatiSyndrome.html.