Dyskeratosis congenital (DC), also called Zinsser–Engman–Cole syndrome, is a rare, multi-system, hereditary disease, which was first described by Zinsser in 1906.1 It is classically diagnosed by the presence of the triad of nail dystrophy, skin reticular pigmentation, and oral leukoplakia. Patients with DC are at a high-risk of bone marrow failure, squamous cell carcinoma of the head and neck, leukemia and other cancers, myelodysplastic syndrome, pulmonary fibrosis, liver disease, and neurological, ophthalmic, genitourinary, and gastrointestinal abnormalities.2,3
Here we report a case of DC without any oral lesion associated with aplastic anemia. Our report emphasizes the fact that not all components of the syndrome need to be present in any given patient, and in all cases of genodermatosis, pleomorphism may be expected with variable clinical manifestations of a single genetic aberration.4 Therefore, physicians should be aware of the mucocutaneous manifestation of DC and of its diagnosis and refer the patient for better evaluation.
Case Report
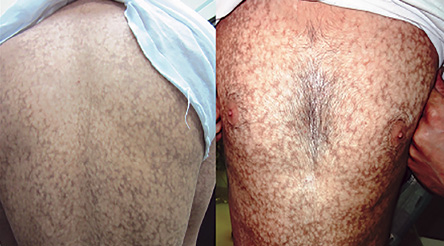
Figure 1: Reticular pigmentation and skin atrophy was seen on the trunk of the patient.
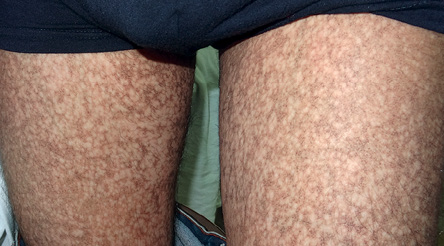
Figure 2: Reticular pigmentation and skin atrophy was visible on both thighs.
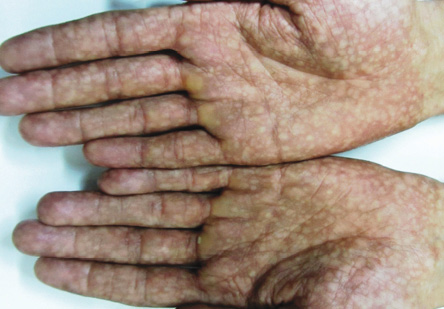
Figure 3: Mottled pigmentation on both palms.
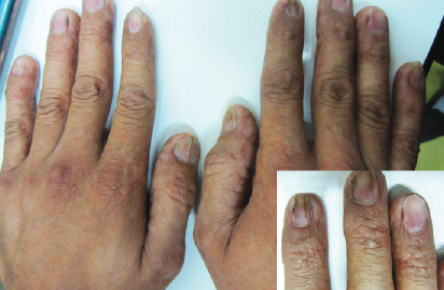
Figure 4: Longitudinal ridging and splitting was seen in some fingernails. Pterygium was visible in the second finger of right hand (insert).
A 27-year-old man was referred to the dermatology clinic of Al-Zahra Hospital, Isfahan University of Medical Sciences (IUMS), Iran, for consultation regarding an asymptomatic reticular pigmentation present on his trunk and extremities for the past nine years. Oral treatment with danazol was considered for treatment of his anemia from three years. The patient did not report itching, photosensitivity, or any other cutaneous symptoms. He was normal until the age of 17. His family history was unremarkable except for aplastic anemia in his brother. The patient appeared pale, but his vital signs were within normal limits, and he showed no other systemic symptoms. Hyperpigmented areas appearing in a reticular pattern were observed on the neck, trunk, and both extremities [Figure 1, 2, and 3]. His fingernails were cracked and atrophied, with some nails having pterygium, which started in late adolescence [Figure 4]. The patient also reported palmar hyperhidrosis. On intraoral examination, no lesions were observed on the tongue or oral cavity. His teeth, periodontium, and other oral mucosal surfaces were normal. Considering the symptoms of skin pigmentation, nail dystrophy, and association with aplastic anemia, DC was considered a probability, and a skin biopsy was performed. Hematological investigations revealed reduced levels of hemoglobin (7mg/dL), platelets, and white blood cells. Because of a lack of facilities for utilization of the cytogenetic method, genetic investigation was not performed.
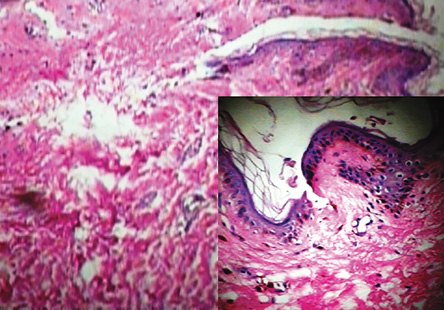
Figure 5: Epidermal atrophy, basal layer degeneration, and melanin incontinence in the dermal papilla of a patient with dyskeratosis congenital. Magnification=400×, insert magnification=100×.
On the basis of the clinical and histopathological findings, and the hematological report, a definitive diagnosis of DC was made. The histopathological report of the reticulated skin lesion revealed mild hyperkeratosis and epidermal atrophy with focal hydropic basal layer degeneration. Focal melanin incontinence was seen in the dermal papilla [Figure 5]. We advised our patient to avoid smoking, severe exposure to the sun, and to return regularly for follow-up.
Discussion
DC normally has a classic triad but in some patients this is not seen. In this disorder, multiple organ systems are affected because of pleiotropic mutation in the gene.5 Abdel-Karim et al,6 reported a variant case of DC affecting a 9-year-old boy where the progression of the disease did not follow the triad pattern and longstanding nail dystrophy and oral ulceration were the sole presenting features in their case. In contrast, Krishnan et al,4 reported a patient who had features consistent with this syndrome including skin atrophy and pigmentation, oral leukoplakia, and esophageal stricture, but with normal fingers and toenails.
Dermatological manifestations occur in the form of reticular hyperpigmentation of the skin. These pigmented areas are associated with atrophy of the epidermis; sun-exposed areas, including the upper trunk, neck, and face, are the most affected.7 Ectodermal abnormalities such as alopecia of the scalp, eyebrows, and eyelashes, premature graying of the hair, hyperhidrosis, and hyperkeratosis of the palms and soles have been noted.8 The same pigmentation was observed in our patient over the skin of the extremities, chest, and back. Oral mucosal changes manifest in the form of a white keratotic patch. Earlier cases reported vesicles and ulcerations preceding the development of leukoplakia.9
Mucosal leukoplakia is a pathognomonic feature and occurs in approximately 80% of patients. It typically involves the buccal mucosa, tongue, and oropharynx. Leukoplakic areas showing an increased risk of malignant transformation, and hence requires frequent monitoring.8 In DC, severe periodontal destruction occurs because of anomalies in ectodermal-derived structures and diminished host response caused by neutropenia.10 Patients have gingival inflammation, bleeding, recession, and bone loss that simulates juvenile periodontitis. There may also be hyperpigmentation of the buccal mucosa that may be reactive postinflammatory hypermelanosis.10 In our case, the patient had no white keratotic patch, vesicles, ulcerations, or any other associated oromucosal abnormality; he had healthy periodontium and teeth.
Dystrophy of the nails is associated with the onset of skin pigmentation. Nail abnormalities can result in longitudinal splitting and furrowing. They become brittle or are completely lost.7 In our patient, the nails of the hand had such features. In DC, eye involvement can lead to epiphora, growth in fundus, blepharitis, and loss of eyelashes.11 Our patient showed none of these symptoms. The majority of patients develop a hematopoietic disorder resembling Fanconi anemia because initially, the bone marrow is normal but, gradually, fat cells and fibrotic tissue replace the hematopoietic cells.9 With the development of pancytopenia, secondary infection occurs, which is the cause of death in the majority of these patients.5 Our patient was pale and had pancytopenia. In his case, aplastic anemia was the first manifestation of the disease. Other abnormalities such as pulmonary fibrosis, alopecia, mental retardation, small genitals, premature aging, nutmeg liver, horseshoe kidneys, amyloidosis, and development of Hodgkin’s disease, and adenocarcinoma have also been reported.3,7,10,12 However, these were not observed in our patient.
To date, there is no effective and curative treatment for DC. Patients should be kept under close observation and recalled for periodic follow-up.13 In these visits, intraoral biopsy and complete blood profiling should be performed to detect any malignant changes in white patches or the development of a hematopoietic disorder.13 Since all symptoms of this disorder are not necessarily seen collectively in any one patient, physicians should consider it a probability in patients showing signs of reticular hyperpigmentation, especially accompanied by aplastic anemia. The physician may be the first to see and diagnose DC and has an important role in the management of this disease. Because of the increased prevalence of malignant mucosal neoplasms, particularly squamous cell carcinoma, increased prevalence of squamous cell carcinoma of the skin, and fatal hematopoietic abnormality, patients should be recalled for regular follow-ups where the physician will have an important role.
Conclusion
Even though many variants have been described in literature, sparing of the oral cavity as in our patient is extremely uncommon. Therefore, physicians should be aware of the mucocutaneous manifestation of DC and its diagnosis and refer the patient for better evaluation.
Disclosure
This study was supported by Skin Disease and Leishmaniasis Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
references
- Zinsser, F. Atrophia cutis reticularis cum pigmentatione dystrophia unguium et leukoplakia oris. Ikonogr Dermal Kioto. 1910;5:219.
- Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am 2009 Apr;23(2):215-231.
- Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood 2009 Jun;113(26):6549-6557.
- Krishnan SS, Yesudian PD, Jayaraman M, Janaki VR, Raj JB. Atypical dyskeratosis congenita. Indian J Dermatol Venereol Leprol 1997 Jan-Feb;63(1):47-49.
- Womer R, Clark JE, Wood P, Sabio H, Kelly TE. Dyskeratosis congenita: two examples of this multisystem disorder. Pediatrics 1983 Apr;71(4):603-609.
- Abdel-Karim A, Frezzini C, Viggor S, Davidson LE, Thornhill MH, Yeoman CM. Dyskeratosis congenita: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2009 Aug;108(2):e20-e24.
- Auluck A. Dyskeratosis congenita. Report of a case with literature review. Med Oral Patol Oral Cir Bucal 2007; 12: E369-73.
- Atkinson JC, Harvey KE, Domingo DL, Trujillo MI, Guadagnini JP, Gollins S, et al. Oral and dental phenotype of dyskeratosis congenita. Oral Dis 2008 Jul;14(5):419-427.
- Loh HS, Koh ML, Giam YC. Dyskeratosis congenita in two male cousins. Br J Oral Maxillofac Surg 1987 Dec;25(6):492-499.
- Treister N, Lehmann LE, Cherrick I, Guinan EC, Woo SB. Dyskeratosis congenita vs. chronic graft versus host disease: report of a case and a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2004 Nov;98(5):566-571.
- Tsilou ET, Giri N, Weinstein S, Mueller C, Savage SA, Alter BP. Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita. Ophthalmology 2010 Mar;117(3):615-622.
- Chakrabarti N, Sarma N, Chattopadhyay C, Chowdhuri AR, Das C, Pal SK. A case of dyskeratosis congenita with primary amenorrhea and adenocarcinoma of stomach. Indian J Dermatol 2011 Sep-Oct;56(5):594-596.
- Handley TP, McCaul JA, Ogden GR. Dyskeratosis congenita. Oral Oncol 2006 Apr;42(4):331-336.