Sex steroids of gonadal and adrenal origin have a critical role in sexual development during the different stages of life from early embryonic development to adulthood. Of the enzymes catalyzing the biosynthesis of these steroid hormones, there are enzymes that are shared in both the adrenal cortex and gonads whose defects cause congenital adrenal hyperplasia, and enzymes that exist mainly in gonads, which are responsible for the synthesis of sex steroids (testosterone in the testes and estrogens in the ovaries).1 Disorders of sex development, including ambiguous genitalia, may develop due to gonadal dysgenesis, androgen biosynthesis defect (17-beta-hydroxysteroid dehydrogenase (17-β-HSD) deficiency or 5α-reductase deficiency), androgen action defect (androgen insensitivity syndrome) or congenital adrenal hyperplasia (21α-hydroxylase, 17α-hydroxylase, 17,20 lyase, 3β-hydroxysteroid dehydrogenase, and cholesterol 20,22 desmolase).1,2
The enzyme 17-β-HSD is part of a group of isoenzymes that are involved in both the synthesis and metabolism of the sex hormones, androgens, and estrogens. 17-β-HSD1 is the major isoenzyme in the ovarian granulosa cells that catalyses the conversion of the less active estrogen, estrone, to the more active estradiol and so promotes maturation of the ovarian follicle. 17-β-HSD2 is an oxidative isoenzyme that inactivates estradiol to estrone and catalyses androgen inactivation in the target tissues. 17-β-HSD3 is expressed almost exclusively in the testis and where it helps to produce the male sex hormone by converting the inactive ∆4C19 steroid androstenedione to the active androgen testosterone. 17-β-HSD4 inactivates both estradiol into estrone and ∆5 androstenediol into dehydroepiandrosterone (DHEA). 17-β-HSD5 catalyzes the formation of testosterone from androstenedione in the peripheral tissues [Figure 1].3,4
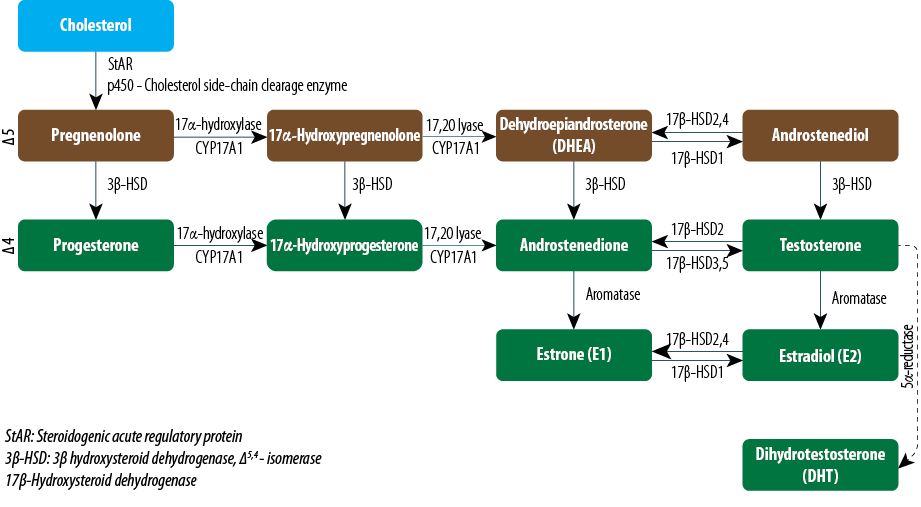
Figure 1: Biosynthesis of steroid hormones in the gonads (testes and ovaries).
17-β-HSD3 deficiency (OMIM: 264300) is a rare autosomal recessive disorder of male sex differentiation (pseudohermaphroditism) resulting from a defect in the last reversible step of steroidogenesis in testosterone biosynthesis in the testes in which androstenedione is converted into testosterone. A mutation in the HSD17B3 gene blocks the synthesis of testosterone in the fetal testis resulting in normal male Wolffian duct structures but with female external genitalia at birth. Homozygous or compound heterozygous 46,XY individuals are characterized by the absence or presence of hypoplastic internal male genitalia (prostate and testes). The diagnosis may be delayed until adolescence in phenotypic females with inguinal hernia, mild clitoromegaly, or urogenital sinus when presented with virilization and primary amenorrhea.5,6
Here we present the first clinically, biochemically, and genetically proven case of androgen biosynthesis defect due to 17-β-HSD3 deficiency in a 46,XY child in Oman and the Gulf region. The diagnosis of this type of pseudohermaphroditism needs a high level of clinical suspicion with proper utilization of laboratory tools for demonstrating the different hormones, their precursors, and metabolites that characterize this disorder with confirmation by molecular mutation analysis.
CASE REPORT
This case report presents the clinical and laboratory course of an 11-year-old boy who was originally referred at the age of six weeks for expert evaluation and management of ambiguous genitalia. He was referred from Nizwa Hospital, a secondary-care regional hospital, to the Royal hospital, a tertiary-care hospital in Oman. The child’s prenatal history was not significant, being born as a full-term baby of a consanguineous parents, through vaginal delivery with good weight. Systemic examination was normal apart from genitalia abnormalities. Examination of the genitalia showed a stretched penile length of 3cm, with undescended testes that were felt bilaterally in the groin. The child was assigned as boy since his delivery. There was no history of similar problems in the family.
At the age of six weeks, laboratory investigations revealed results within the reference ranges for core blood tests including electrolytes, renal, liver, bone, glucose, and thyroid profiles. The results for adrenal and gonadal steroids, pituitary hormones, and karyotyping are shown in Table 1. The low testosterone and androstenediol glucuronide, high androstenedione and estrone levels are consistent with testosterone synthetic defect due to 17-β-HSD3 deficiency. Urine steroid profile revealed a normal quantitative pattern, which excludes 5α-reductase deficiency, which usually has comparable presentation. Ultrasound of the groin and pelvis showed no obvious evidence of any uterine or ovarian structures. There were oval shaped solid structures in both inguinal regions, each measuring about 1.5cm, which looked like testicular structures. A biopsy was performed and the histology report revealed immature seminiferous tubules only. No ovarian tissues were seen. He was referred to pediatric surgery for surgical correction of the hypospadias.
Table 1: Plasma steroids and pituitary hormones at time of presentation at the age of six weeks (compared to recommended age and sex matched reference ranges, if age and sex dependent).
|
Testosterone (nmol/L) |
2.5 |
0.0–6.5
(2–5 weeks) |
|
Luteinizing hormone
(IU/L) |
32.6 |
0.3–2.8
(2 weeks–10 years) |
|
Follicle-stimulating
hormone (IU/L) |
7.0 |
0–2.5
(2 weeks–3 years) |
|
Androstenedione
(nmol/L) |
12.6 |
<1.7 |
|
Androstanediolglucuronide (nmol/L) |
6.0 |
8.5–80 |
|
Dehydroepiandrosterone
sulfate (umol/L) |
1.0 |
0.2–8.6
(≤1 month) |
|
Estrone (pmol/L) |
62.9 |
<40
(prepubertal range) |
|
Estradiol (pmol/L) |
109 |
37–117 (prepubertal range) |
|
Progesterone(nmol/L) |
2.1 |
2.7–10.7
(0–12 months) |
Based on the laboratory finding, the child was diagnosed as having 17-β-HSD3 deficiency. During the course of his management, testosterone injections (25mg once monthly) were given: at two and six months then at three months before his surgeries at the age of five and seven years when he underwent multiple operations for orchidopexy and hypospadias correction. He showed good response that was reflected in terms of an increase in phallas length.
Patient was followed-up in the Pediatric Endocrinology Clinic annually, until the age of 10 years when he presented with gynecomastia that was progressive and associated with voice change as well as bad body odor. On examination, his weight was 41.8kg (which had increased from the previous year by approximately 10kg), height was 141.9cm, and BMI was 22kg/m2. On examination the gynecomastia was bilateral, stage 4, with pubic hair Tanner 3 (p3) and testicular volume 8ml. Laboratory investigations showed normal serum liver profile, beta-human chorionic gonadotropin (β-HCG) and α-fetoprotein levels, which excluded other causes of gynecomastia. He had also raised follicle-stimulating hormone (FSH), luteinizing hormone (LH), androstenedione, and estrone with low-normal testosterone, low androstendiol glucurunide, and normal DHEA levels [Table 2]. At this age, wrist x-ray examination showed a normal bone age compared with his chronological age. The magnetic resonance imaging showed normal pituitary gland and hypothalamic structures with no brain abnormality.
Table 2: Plasma steroids and pituitary hormones at age of 10 years (compared with recommended age and sex matched reference ranges).
|
Testosterone (nmol/L) |
3.6 |
0.1–2.4 (prepubertal)
9.0–31.0 (adult) |
|
Luteinizing hormone
(IU/L) |
25.2 |
1.5–9.0 |
|
Follicle-stimulating
hormone (IU/L) |
59.3 |
1–12 |
|
Androstenedione (nmol/L) |
24.4 |
<1.7 |
|
Androstanediolglucuronide (nmol/L) |
6.0 |
8.5–80 |
|
Dehydroepiandrosterone
sulfate (umol/L) |
2.88 |
0.5–6.6 |
|
Estrone (pmol/L) |
280 |
30–220 |
|
Estradiol (pmol/L) |
66.0 |
20–95 (Tanner 3) |
The gynecomastia could be explained by 17-β-HSD3 deficiency and the high FSH and LH levels, which indicated primary gonadal failure. As this patient had significant gynecomastia with low testosterone, he was given testosterone injections, 50mg once a month, for a total of six months. He was followed-up and reviewed three months after commencing testosterone therapy. There was a significant reduction in his gynecomastia with much improvement in his psychological status. He remains under follow-up every six months to check for virilization during puberty, which is expected in his case. His fertility status will also be evaluated in the future as he is at risk of infertility.
Molecular genetic analysis
DNA was extracted from the patient’s peripheral blood sample using QIAsymphony kit (Qiagen, USA). All 11 exons of the HSD17B3 gene were analyzed using the polymerase chain reaction (PCR) and sequencing of both DNA strands of the entire coding region was carried out, including the highly conserved exon-intron splice junctions. The HSD17B3 gene provides instructions for making an enzyme called 17-β-hydroxysteroid dehydrogenase 3. Mutations in the HSD17B3 gene result in a 17-β-hydroxysteroid dehydrogenase 3 enzyme with little or no activity, thus reducing testosterone production.
The sequencing analysis revealed a previously unreported homozygous variant in exon 8 of the HSD17B3 gene (c.576G>A.Trp192*) [Figure 2]. This variant creates a premature stop codon, which is likely to result in a truncated protein or loss of protein production. Parental samples were not available in order to confirm the homozygosity in place of compound heterozygosity for a large deletion. However, given the parent’s consanguinity (they are first cousins) and the fact that so far no large deletion/duplication has been described in the HSD17B3 gene7 being homozygous is the most likely scenario for the detected mutation.
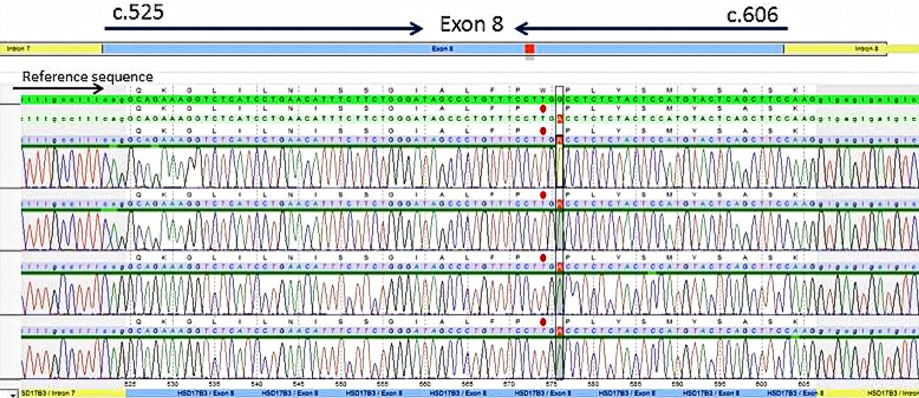
Figure 2: Electropherogram of sequencing data of exon eight of the HSD17B3 gene (NM_000197.1).
DISCUSSION
This is the first case of 17-β-HSD3 deficiency to be reported in Oman and the Gulf region. The clinical and laboratory course of an 11-year-old boy diagnosed with 17-β-HSD3 deficiency at the age of six-weeks old following his presentation with ambiguous genitalia (stretched penile and bilateral undescended testes) is described. The clinical, imaging and laboratory findings (adrenal and gonadal steroids, pituitary hormones, and karyotype) were consistent with 17-β-HSD3 deficiency. The diagnosis was confirmed by genetic analysis due to presence of a novel homozygous variant in exon eight of the HSD17B3 gene (c.576G>A.Trp192*). This nonsense variant creates a premature stop codon, which is very likely to result in a truncated protein or loss of protein production. This novel mutation has not been described so far in the literature for the HSD17B3 gene.
The gene HSD17B3, encoding 17β-HSD3, contains 11 exons and has been cloned and mapped to chromosome 9q22. To date, 35 mutations have been reported in the HSD17B3 gene, three of which are STOP mutations.7 Although mutations throughout the gene have been described, a mutation cluster region in exon nine with complete elimination of 17β-HSD3 activity was identified in many populations.3,5,8-15 Moreover, so far, only three pathogenic mutations have been reported in patients with Middle Eastern ancestry as follows:
- P.R80Q mutation in exon 3 that has been identified in Palestinian, Turkish, Iranian and Brazalian populations.16-18 Roster et al,14 were the first to identify this mutation in 24 subjects from nine Arab families from Gaza, Jerusalem, Lod, and Ramle.5,10
- A novel homozygous splice-site mutation (c.524 + 2T>A) in intron 7.
- A novel homozygous missense mutation in exon 11 with premature stop codon (p. Y287*), described in 46,XY Sudanese and Turkish, phenotypic females who presented with primary amenorrhea and virilism respectively.18,19
17-β-HSD3 deficiency is a genetic steroid disorder of testicular androgen synthesis that was first described by Saez et al.19 As in our case, male newborns with 17-β-HSD3 deficiency usually have external genitalia with feminizing features together with undescended testes usually in the inguinal region or in a bifid scrotum.5,10,11 The presence of Wolffian duct structures such as epididymis, seminal vesicles, vas deferens, and ejaculatory ducts may be explained by the low testosterone concentration, which appears to be sufficient for their development in utero. In addition, testosterone production through an alternative pathway catalyzed by other 17-β-HSD isoenzymes may contribute to the androgenization of these structures.5,6
The laboratory diagnosis of 17-β-HSD3 deficiency is usually made based on finding a characteristic biochemical pattern with predominance in 17-ketosteroids (namely androstenedione, DHEA, and estrone) compared with 17-hydroxylsteroids (namely testosterone, androstendiol, and estradiol) with consequent increase in androstenedione:testosterone and estrone:estradiol ratios in basal state or post-HCG stimulation. This biochemical profile was demonstrated in our case since six weeks of age when the diagnosis was made. The urine steroid profile was normal, which excludes 5α-reductase deficiency that usually has comparable presentation. Confirmation of the diagnosis and mutation type was done at the prepubertal age due to the importance of this age on virilization and fertility state.1,5,6,9,11
The decision for sex rearing in patients with 17-β-HSD3 deficiency is difficult especially that the majority of cases are diagnosed late in childhood or at puberty. Also, consensus guidelines do not clearly support one gender assignment although there is more support to male gender.20 Sex rearing is usually revisited in these patients at the time of puberty when they develop marked virilization with penis enlargement, male pattern body hair, and muscle development.12,13 Puberty-dependent virilization pushes many patients to change their social sex to male at puberty whether with or without surgical correction, otherwise they require bilateral orchiectomy if the female social sex is chosen. The consequent female-to-male gender reassignment has been reported in 39–64% of cases.21 However, almost all diagnosed cases of Arab patients involve male social sex assignment.15,17,21-26
The excessive virilization at puberty and its discrepancy from intrauterine masculinization is not fully understood, but it is thought to occur by one of two mechanisms. The first is attributed to the peripheral conversion of androstenedione to testosterone by other 17-β-HSD isoenzymes particularly isoenzyme five in extragonadal tissues. The second mechanism is attributed to the raised LH in patients with 17-β-HSD3 deficiency, which in turn increases testicular testosterone production in patients with residual 17-β-HSD3 function.3 In addition, the expression of aldo-keto reductase family 1 member C3 (AKR1C3; 17-β-HSD5) has been demonstrated in extragonadal tissues in response to high LH in both normal subjects and patients with 17-β-HSD3 deficiency.27 The high concentration of androstenedione is converted to testosterone by the cells containing AKR1C3 (17-β-HSD5) which include extra gonadal tissues such as genital skin and adipose tissue as well as the Leydig cells of 17-β-HSD3 deficient patients.28,29 Bilateral orchiectomy has been reported to result in a marked decrease in androstenedione as well as in virilization confirming the role of the testis as the main source of testosterone production in these patients.3,30
Conclusion
We report the first genetically proven case of 17-β-HSD3 deficiency in Oman and the Gulf region. The diagnosis is usually made by demonstrating a characteristic biochemical pattern in a profile of gonadal steroid levels together with confirmation by molecular testing. Mutation analysis in our case revealed a novel homozygous variant in exon 8 of the HSD17B3 gene (c.576G>A.Trp192*), which is the first to be reported in the literature.
Disclosure
The authors declared no conflicts of interest.
Acknowledgements
The authors would like to acknowledge Centogene Laboratory (Centrogene AG, Schillingallee 68, 18057 Rostock, Germany), for performing the molecular testing for this patient.
references
- Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 2011 Feb;32(1):81-151.
- Mula-Abed WA, Pambinezhuth FB, Al-Kindi MK, Al-Busaidi NB, Al-Muslahi HN, Al-Lamki MA. Congenital adrenal hyperplasia due to 17-alpha-hydoxylase/17,20-lyase deficiency presenting with hypertension and pseudohermaphroditism: First case report from Oman. Oman Med J 2014 Jan;29(1):55-59.
- Andersson S, Moghrabi N. Physiology and molecular genetics of 17 beta-hydroxysteroid dehydrogenases. Steroids 1997 Jan;62(1):143-147.
- Labrie F, Luu-The V, Lin SX, Labrie C, Simard J, Breton R, et al. The key role of 17 beta-hydroxysteroid dehydrogenases in sex steroid biology. Steroids 1997 Jan;62(1):148-158.
- George MM, New MI, Ten S, Sultan C, Bhangoo A. The clinical and molecular heterogeneity of 17βHSD-3 enzyme deficiency. Horm Res Paediatr 2010 Aug;74(4):229-240.
- Prehn C, Möller G, Adamski J. Recent advances in 17beta-hydroxysteroid dehydrogenases. J Steroid Biochem Mol Biol 2009 Mar;114(1-2):72-77.
- HGMD. Human Genome Mutation Database; 2014. Version 4. Available at https://portal.biobase-international.com/hgmd/pro/gene.php?gene=HD17B3
- Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonca BB, et al. Male pseudohermaphroditism caused by mutations of testicular 17 beta-hydroxysteroid dehydrogenase 3. Nat Genet 1994 May;7(1):34-39.
- Andersson S, Geissler WM, Wu L, Davis DL, Grumbach MM, New MI, et al. Molecular genetics and pathophysiology of 17 beta-hydroxysteroid dehydrogenase 3 deficiency. J Clin Endocrinol Metab 1996 Jan;81(1):130-136.
- Boehmer AL, Brinkmann AO, Sandkuijl LA, Halley DJ, Niermeijer MF, Andersson S, et al. 17Beta-hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. J Clin Endocrinol Metab 1999 Dec;84(12):4713-4721.
- Mendonca BB, Domenice S, Arnhold IJ, Costa EM. 46,XY disorders of sex development (DSD). Clin Endocrinol (Oxf) 2009 Feb;70(2):173-187.
- Rosler A. 17 beta-hydroxysteroid dehydrogenase 3 deficiency in the Mediterranean population. Pediatr Endocrinol Rev 2006 Aug;3(3)(Suppl 3):455-461.
- Rösler A, Kohn G. Male pseudohermaphroditism due to 17 beta-hydroxysteroid dehydrogenase deficiency: studies on the natural history of the defect and effect of androgens on gender role. J Steroid Biochem 1983 Jul;19(1B):663-674.
- Rösler A, Silverstein S, Abeliovich D. A (R80Q) mutation in 17 beta-hydroxysteroid dehydrogenase type 3 gene among Arabs of Israel is associated with pseudohermaphroditism in males and normal asymptomatic females. J Clin Endocrinol Metab 1996 May;81(5):1827-1831.
- Bertelloni S, Balsamo A, Giordani L, Fischetto R, Russo G, Delvecchio M, et al. 17beta-Hydroxysteroid dehydrogenase-3 deficiency: from pregnancy to adolescence. J Endocrinol Invest 2009 Sep;32(8):666-670.
- Bilbao JR, Loridan L, Audí L, Gonzalo E, Castaño L. A novel missense (R80W) mutation in 17-beta-hydroxysteroid dehydrogenase type 3 gene associated with male pseudohermaphroditism. Eur J Endocrinol 1998 Sep;139(3):330-333.
- Omrani MD, Adamovic T, Grandell U, Saleh-Gargari S, Nordenskjöld A. 17-β-hydroxysteroid dehydrogenase type 3 deficiency in three adult Iranian siblings. Sex Dev 2011 Dec;5(6):273-276.
- Shammas C, Neocleous V, Toumba M, Costi C, Phedonos AA, Efstathiou E, et al. Overview of genetic defects in endocrinopathies in the island of Cyprus; evidence of a founder effect. Genet Test Mol Biomarkers 2012 Sep;16(9):1073-1079.
- Saez JM, De Peretti E, Morera AM, David M, Bertrand J. Familial male pseudohermaphroditism with gynecomastia due to a testicular 17-ketosteroid reductase defect. I. Studies in vivo. J Clin Endocrinol Metab 1971 May;32(5):604-610.
- Cohen-Kettenis PT. Gender change in 46,XY persons with 5alpha-reductase-2 deficiency and 17beta-hydroxysteroid dehydrogenase-3 deficiency. Arch Sex Behav 2005 Aug;34(4):399-410.
- Lee YS, Kirk JM, Stanhope RG, Johnston DI, Harland S, Auchus RJ, et al. Phenotypic variability in 17beta-hydroxysteroid dehydrogenase-3 deficiency and diagnostic pitfalls. Clin Endocrinol (Oxf) 2007 Jul;67(1):20-28.
- Ellaithi M, Werner R, Riepe FG, Krone N, Kulle AE, Diab T, et al. 46,XY disorder of sex development in a sudanese patient caused by a novel mutation in the HSD17B3 gene. Sex Dev 2014 Aug;8(4):151-155.
- Houk CP, Hughes IA, Ahmed SF, Lee PA; Writing Committee for the International Intersex Consensus Conference Participants; International Intersex Consensus Conference. Summary of consensus statement on intersex disorders and their management. Pediatrics 2006 Aug;118(2):753-757.
- Mains LM, Vakili B, Lacassie Y, Andersson S, Lindqvist A, Rock JA. 17beta-hydroxysteroid dehydrogenase 3 deficiency in a male pseudohermaphrodite. Fertil Steril 2008 Jan;89(1):228.e13-7.
- Alikasifoglu A, Hiort O, Gonc N, Demirbilek H, Isik E, Kandemir N. 17beta-hydroxysteroid dehydrogenase type 3 deficiency as a result of a homozygous 7 base pair deletion in 17betaHSD3 gene. J Pediatr Endocrinol Metab 2012;25(5-6):561-563.
- Ben Rhouma B, Belguith N, Mnif MF, Kamoun T, Charfi N, Kamoun M, et al. A novel nonsense mutation in HSD17B3 gene in a Tunisian patient with sexual ambiguity. J Sex Med 2013 Oct;10(10):2586-2589.
- Werner R, Kulle A, Sommerfeld I, Riepe FG, Wudy S, Hartmann MF, et al. Testosterone synthesis in patients with 17β-hydroxysteroid dehydrogenase 3 deficiency. Sex Dev 2012 Jun;6(4):161-168.
- Ashley RA, Yu Z, Fung KM, Frimberger D, Kropp BP, Penning TM, et al. Developmental evaluation of aldo-keto reductase 1C3 expression in the cryptorchid testis. Urology 2010 Jul;76(1):67-72.
- O’Reilly MW, House PJ, Tomlinson JW. Understanding androgen action in adipose tissue. J Steroid Biochem Mol Biol 2014 Sep;143:277-284.
- Mendonca BB, Inacio M, Arnhold IJ, Costa EM, Bloise W, Martin RM, et al. Male pseudohermaphroditism due to 17 beta-hydroxysteroid dehydrogenase 3 deficiency. Diagnosis, psychological evaluation, and management. Medicine (Baltimore) 2000 Sep;79(5):299-309.
- Tuhan HU, Anik A, Catli G, Ceylaner S, Dundar B, Bober E, et al. A novel missense mutation in HSD17B3 gene in a 46, XY adolescent presenting with primary amenorrhea and virilization at puberty. Clin Chim Acta 2015 Jan;438(8):154-156.