|
Abstract
Advances in the identification of gastrointestinal stromal tumors, its molecular and immunohiostochemical basis, and its management have been a watershed in the treatment of gastrointestinal tumors. This paradigm shift occurred over the last two decades and gastrointestinal stromal tumors have now come to be understood as rare gastrointestinal tract tumors with predictable behavior and outcome, replacing the older terminologies like leiomyoma, schwannoma or leiomyosarcoma. This report presents a case of gastric gastrointestinal stromal tumor operated recently in a 47-year-old female patient and the outcome, as well as literature review of the pathological identification, sites of origin, and factors predicting its behavior, prognosis and treatment.
Keywords: Gastrointestinal stromal tumor; Identification; Risk stratification; Management.
Introduction
Gastrointestinal stromal tumors (GISTs), though the most common mesenchymal tumors of the GI tract, are rare accounting approximately 1% to 3% of all gastrointestinal tumors. Mazur and Clarke coined the term GIST in 1983 for a distinct set of mesenchymal tumors of the GI tract having no ultrastructural or immunohistochemical features characteristic of smooth muscle differentiation.1
Kindblom and associates in 1998 demonstrated that the actual cell of origin of these tumors is a pluripotent mesenchymal stem cell programmed to differentiate into interstitial cells of Cajal, the GI tract "pacemaker cells" - the cells responsible for initiating and co-coordinating GI motility.2 Perhaps, the most critical development that distinguished GIST as a unique clinical entity was the discovery of c- kit proto-oncogene gain-of-function mutations in these tumors by Hirota and collegues in 1998.3
These advances led to the development of molecular targeted therapy for adjuvant and neoadjuvant protocols, using tyrosine kinase inhibitors like imatinib mesylate. These agents act by competing for the ATP binding site on the target kinase, inhibiting tyrosine kinase and reducing cellular proliferation.
Case Report
This is a recent case of an exophytic GIST of gastric origin. A 47-year-old female presented with vague upper abdominal pain and feeling of an abdominal lump on and off for two years. There were no other associated general or GI tract symptoms. Clinical examination of the abdomen revealed a well-defined transversely mobile intra-abdominal lump in the right hypochondrium of about 8 × 6 cm.
USG and CECT of the abdomen reported a large tumor arising probably from the greater curve of the stomach or omentum displacing the transverse colon caudally. (Figs. 1, 2, and 3)
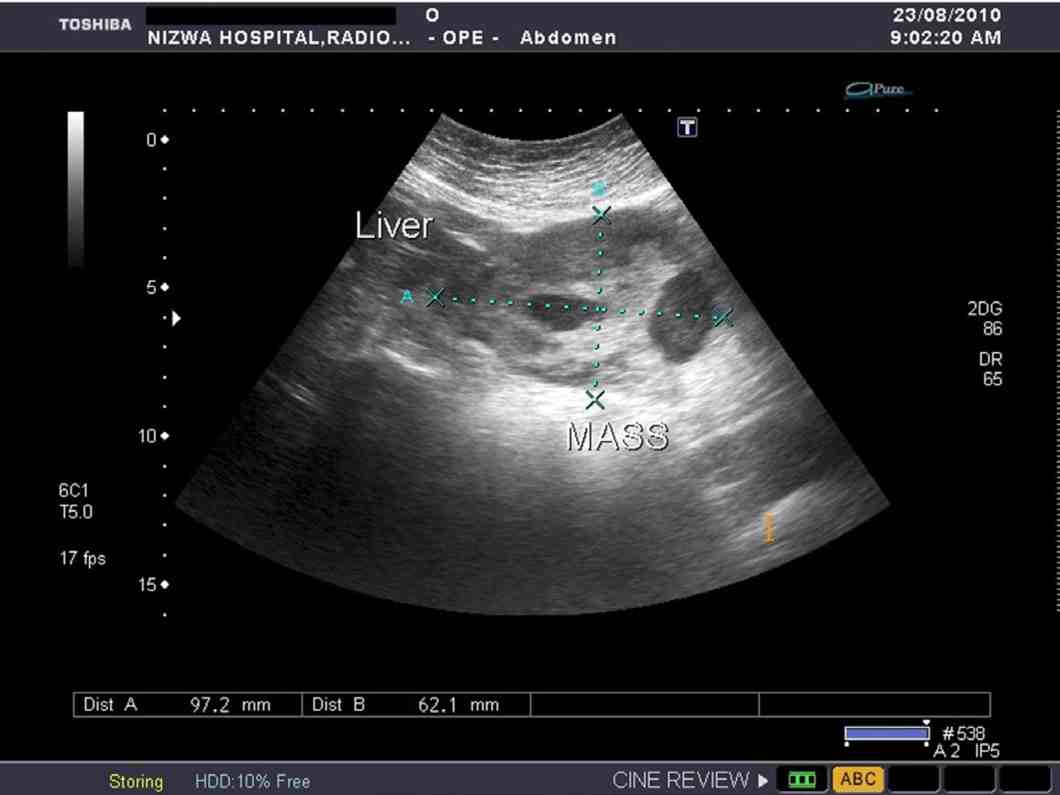
Figure 1: USG Abdomen showing a solid heterogenous mass with necrotic areas, lateral to the left lobe of the liver.
The abdomen was explored electively and a 10 × 6 cm tumor was seen arising from the greater curve of the stomach, exophytically, with a sessile base. There was no infiltration of the mass into the surrounding structures, nor any evidence of metastases or lymphadenopathy. The tumor was excised with a 2 cm sleeve of the greater curve of the stomach and the latter repaired classically. (Fig. 4)
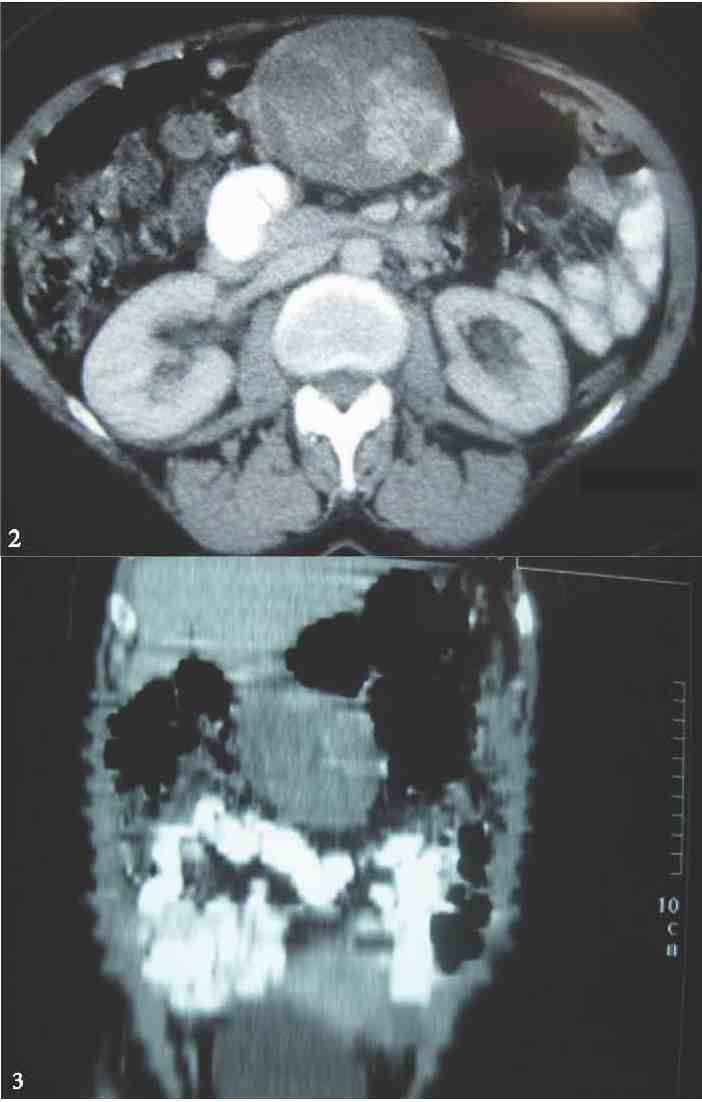
Figures 2 & 3: Axial section and Coronal reformat from CECT Abdomen showing a solid mass with heterogenous contrast enhancement, arising from the greater curvature / omentum displacing the transverse colon caudally. No definite infiltration of surrounding structures.
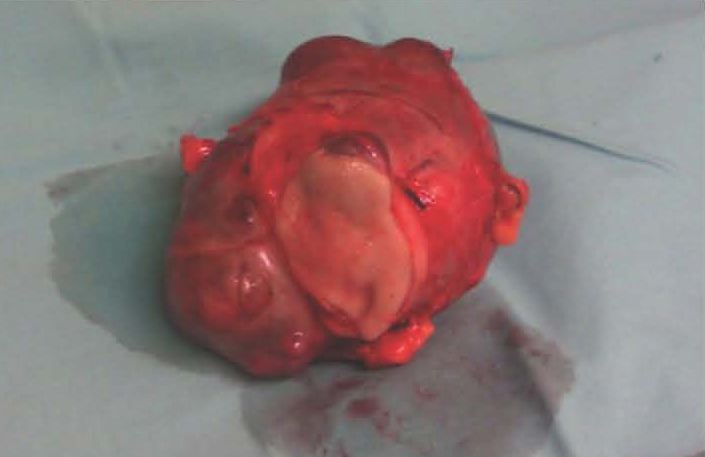
Figure 4: Excised tumor.
Histopathology of the tumor revealed a 9 × 5.4 × 5 cm sized neoplasm arising from the gastric submucosa composed of fusiform and epitheloid cells. Mitosis was occasional at 2/50 hpf. Immunohistochemistry reported that the tumor cells were positive for vimentin, CD 34, S-100, SMA, and desmin. PanCK and LCA were negative. The findings favored the diagnosis of epitheloid gastrointestinal stromal tumor of low malignant potential. The resected margin was reported clear of the tumor. The risk stratification in relation to the location, size and number of mitosis of the tumor revealed a 3.6% chance of disease progression, which is a low risk category.
The patient was discharged in a stable condition. She has been on follow-up for 36 months postoperatively. Serial ultrasonogram and CECT of the abdomen has been reported to be normal with no evidence of recurrence. Adjuvant imatinib therapy was not considered as the prognostic factors revealed a low risk. She is scheduled for a long term followup both under the General Surgery and Medical Oncology Departments.
Discussion
Gastrointestinal stromal tumor can occur anywhere in the GI tract. They are submucosal lesions frequently growing endophytically. They also manifest exophytically. Sizes of these tumors have been reported from small 1 cm to large 40 cm diameter excrescences. About 50% to 75% of these originate in the stomach and about 20% in the small bowel, while less frequent sites include the colon and Rectum. Brunner's gland hamartomas of the duodenum mimic the radiological and endoscopic features of GIST.4
GIST occurs marginally frequent in males as compared to females, both in the fifth and sixth decades of life. There is no racial or geographical preponderance. Clinical presentation varies from an incidental radiological finding when a patient is investigated for other symptoms to cases of intestinal obstruction, upper or lower GI bleeding or melena, and also as an emergent idiopathic spontaneous intra-abdominal hemorrhage. As in the case being reported, some may present with a palpable abdominal lump.
Although abdominal ultrasound is often the initial imaging test employed in the investigation of a patient with abdominal pain or mass, the tumor discovered is frequently so large rendering the organ of origin unidentifiable. The sonogram report frequently indicates the presence of a huge mass, often filling the abdomen, of heterogeneous reflectivity and frequent necrosis. CT therefore provides the basis for diagnosis and staging in most patients. Tumors are usually of varying density, and show patchy enhancement after intravenous contrast. Varying degrees of necrosis may be frequently demonstrated within the mass.5 The CT study will usually provide rapid and reproducible assessment of the size of the primary tumor, as well as its relationship to other structures. Metastatic disease may well be demonstrated at the outset.6 CECT abdomen findings of solid pseudopapillary neoplasm of the pancreas could share the radiological features of GIST.7
GIST can develop into an enormous size before diagnosis and demonstrate considerable cystic change usually associated with a surrounding rim of the viable enhancing tumor. Necrosis may lead to enteric fistulation. Calcification within the tumor is occasionally recognized in association with this tumor necrosis.
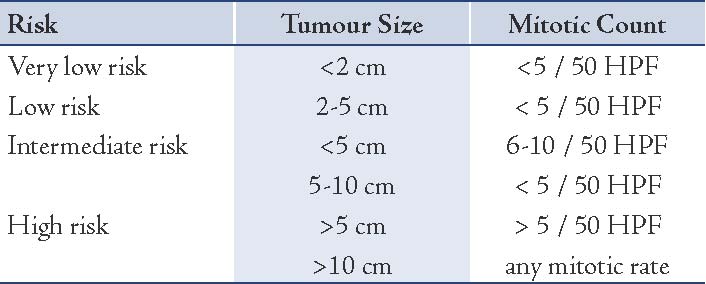
The curative intent in the treatment is operative excision with a clear margin, R0. The prognosis initially rested upon the risk factors of the size of the tumor and its mitotic activity. The high risk of malignant potential and recurrence are seen in tumors more than 5-10 cm in size with a mitotic count of more than 10/50 hpf. Recurrences reach up to 80% in these high risk groups. One of the earliest schemes of predicting risk factors was stratified by Fletcher et al. in 2002.8 (Table 1)
Table 1: A scheme of predicting risk factors.8
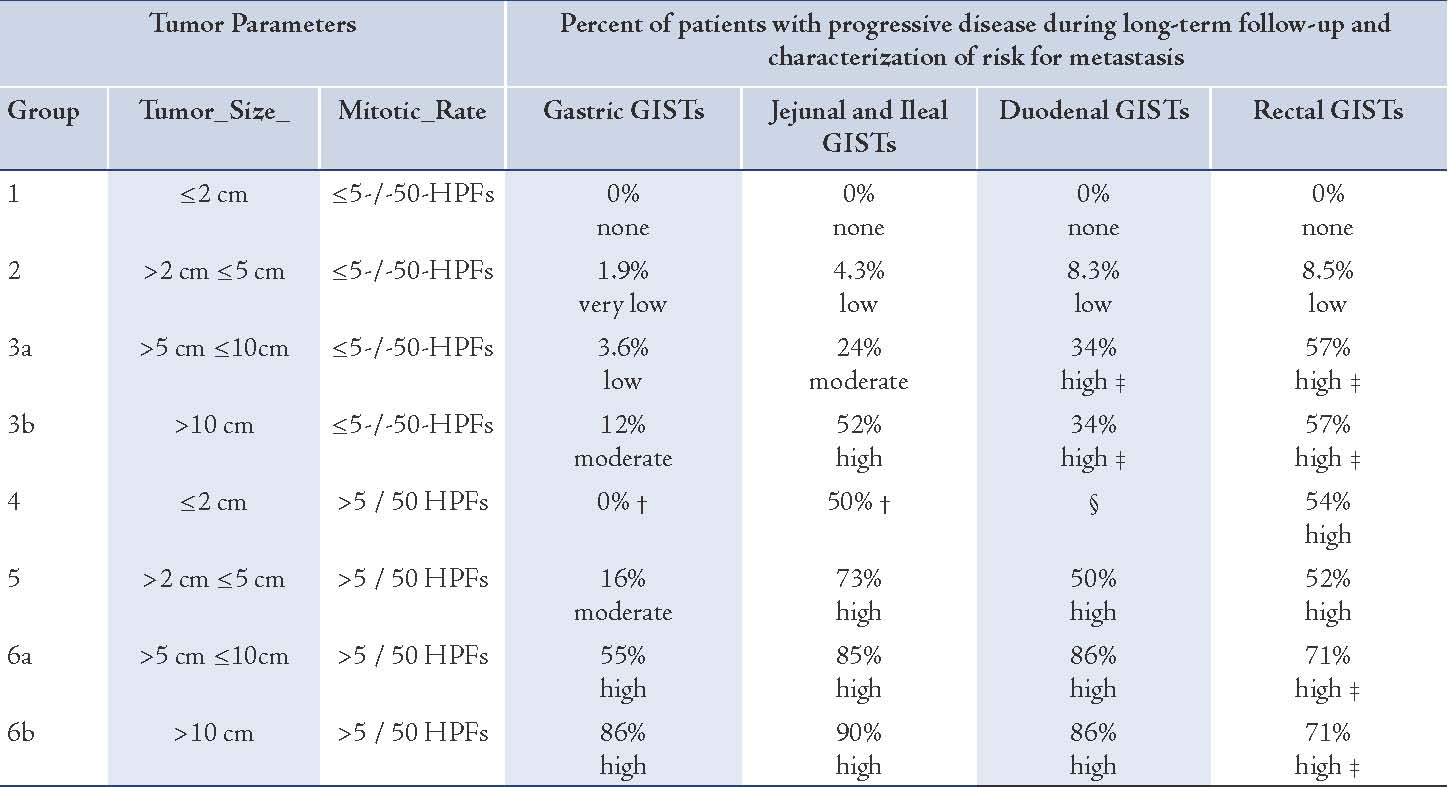
Nilsson et al. (2005) gathered data up to the year 2000 for all c-KIT positive GIST in Sweden. As seen in Fletcher’s Risk Factors, 62.5% of cases of the High Risk Group showed recurrence or metastases.9 Nakamura et al. in 2005 also found 38.5% cases of recurrence and metastases in the high risk group. These studies confirmed that the risk scheme by Fletcher was a good predictor of recurrences and metastases.10 Numerous authors have found a relationship between the risk of recurrence and metastases showing variance to the anatomical location of the primary GIST. Miettinen and Lasota in 2006 have refined the risk table on follow- up information of 1900 patients having GIST over time.11 (Table 2)
Table 2: Risk Table on follow up information.11
In emergent situations and presentations, the acute life-threatening clinical pictures of intestinal obstruction, upper or lower GI bleeding, or idiopathic spontaneous intra-abdominal hemorrhage are dealt with. The mainstay of treatment is surgical excision having a clear margin. Traditional Chemotherapy and Radiotherapy have not been found to be effective.
The consensus guidelines for management of GIST by the ESMO and ASCO groups are outlined as follows:
Small esophagogastric or duodenal nodules <2 cm are deemed as low risk and these patients need only follow-up, reserving excision for patients whose tumor increase in size or become symptomatic.
Standard approach for nodules >2 cm is excision biopsy. If larger and surgery is expected to involve multivisceral resection, multiple core needle biopsies are performed guided by ultrasonography or CT.
Neoadjuvant imatinib is considered for 6-12 months to achieve cytoreduction if R0 excision is not considered possible or the surgery entails gross functional sequelae. Tumor response is assessed by serial ultrasonogram or CT. The risk of relapse is estimated on the basis of mitotic rate, tumor size, tumor site, surgical margins and whether tumor ruptured during excision. Spontaneous tumor rupture or during excision denotes a high risk independent of any other prognostic factor.12 Staging procedures relate to the fact that most metastases or recurrences affect the peritoneum and liver. Ultrasonogram, CT abdomen and PET scans are recommended, the latter, useful to detect tumor response to neoadjuvant or adjuvant targeted molecular therapy.13
With the advent of imatinib mesylate since 2000, targeted molecular therapy has made successful inroads in the management of patients with operated GIST having no clear margins, tumors which are unresectable, or those with recurrences. This drug, along with the recently introduced sunitinib has been used with promising results over the last few years for adjuvant and neoadjuvant therapies. The drug was approved by the FDA for use against metastatic GIST in 2001 and for prevention of recurrences in operated GIST in 2008, for intermediate and high risk groups.
Imatinib mesylate is a multitargeted c-KIT, PDGF–R, and c-ABL inhibitor ensuring arrest of T–cell proliferation. Imatinib binds preferentially to ATP binding sites of c-KIT proto-oncogene product, platelet-derived growth factor receptor (PDGF-R), and Abelson Kinase (c-ABL), impeding the ensuing signal transduction.
Use of adjuvant imatinib mesylate has successfully increased the overall survival (OS) and progression free survival (PFS). Some patients develop resistance to Imatinib during long-term therapy. The acquired resistance to the drug occurs commonly via a secondary mutation in the KIT kinase domain, located in exon 17. Mutations in exon 9 and 11 have been reported. KIT exon 9 mutations seem to define a distinct subset of GIST located in the small bowel and associated with an unfavorable outcome.14
Mutational analysis and testing is an evolving concept. Although mutational analysis has become routine in many academic centers, standard guidelines for its clinical significance is yet to be established. GIST with exon 11 mutations appears to be more sensitive to imatinib. The tumors with exon 9 mutations may benefit from higher doses of imatinib. Such patients whose disease progressed on imatinib therapy may be more sensitive to sunitinib. The novel oral multikinase inhibitor, regorafenib, appears to provide a significant improvement in progression free survival in patients with metastatic or unresectable GIST after failure of imatinib and sunitinib as evidenced by the GRID (GIST-Regorafenib in Progressive Disease) Study in its Phase III trial of the drug.
Guidelines indicate that radical surgical resection is the gold standard for localized primary GIST. Increasing cure rates, overall survival and progression-free survival should be the aim of all adjuvant therapy which should be reserved only for patients having significant prognostic indicators for disease recurrence.15 The Scandinivian Sarcoma Group study showed that adjuvant imatinib given for three years improved recurrence-free survival compared to a one-year therapy.
Recent studies have shown the risk of recurrence is high if tumor spillage occurs during surgery. The present consensus is that patients who have histological profile of intermediate, moderate, or high risk and those with R1 and R2 (microscopic and macroscopic tumor residue) or tumor rupture should receive long-term adjuvant therapy with imatinib. Nomograms have been developed to predict recurrence-free survival after resection of localized primary GIST tumors. This helps to guide patient selection for adjuvant imatinib therapy.16
Conclusion
Awareness of GIST as a distinct GI tract lesion is paramount in managing these rare and often aggressive tumors, which, given the risk factors now known, show a significant preponderance for recurrence and behave as a malignant lesion. A successful outcome requires a multidisciplinary approach, postoperative targeted molecular therapy in intermediate and high risk patients, and continued surveillance.
Acknowledgements
The authors reported no conflict of interest and no funding was received for this work.
References
1. Mazur MT, Clark HB. Gastric Stromal Tumours: Reappriasal of Histogenesis. Am J Surg Pathol 1983;•••:7509-7519.
2. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998 May;152(5):1259-1269.
3. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998 Jan;279(5350):577-580.
4. Gokhale U, Pillai GR. Large Brunner’s Gland Hamartoma: A Case Report. Oman Med J 2009 Jan;24(1):41-43.
5. King DM. The radiology of gastrointestinal stromal tumours (GIST). Cancer Imaging 2005;5(1):150-156.
6. Burkill GJ, Badran M, Al-Muderis O, Meirion Thomas J, Judson IR, Fisher C, et al. Malignant gastrointestinal stromal tumor: distribution, imaging features, and pattern of metastatic spread. Radiology 2003 Feb;226(2):527-532.
7. Lakhtakia R, Al-Wahaibi K, Zahid KF, Malik KA, Burney IA. Solid pseudopapillary neoplasm of the pancreas: a case report with review of the diagnostic dilemmas and tumor behavior. Oman Med J 2013 Nov;28(6):441-444.
8. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol 2002 May;33(5):459-465.
9. Nilsson B, Bümming P, Meis-Kindblom JM, Odén A, Dortok A, Gustavsson B, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era–a population-based study in western Sweden. Cancer 2005 Feb;103(4):821-829.
10. Nakamura N, Yamamoto H, Yao T, Oda Y, Nishiyama K, Imamura M, et al. Prognostic significance of expressions of cell-cycle regulatory proteins in gastrointestinal stromal tumor and the relevance of the risk grade. Hum Pathol 2005 Jul;36(7):828-837.
11. Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001 Jan;438(1):1-12.
12. Hohenberger P, Ronellenfitsch U, Oladeji O, Pink D, Ströbel P, Wardelmann E, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumour. Br J Surg 2010 Dec;97(12):1854-1859.
13. Blackstein ME, Corless CL, Ballman KV, et al. Risk assessment for tumor recurrence after surgical resection of localized primary gastrointestinal stromal tumor (GIST): North American Intergroup phase III trial ACOSOG Z9001. Program and abstracts of the 2010 Gastrointestinal Cancers Symposium; January 22-24, 2010; Orlando, Florida. Abstract 6.
14. Sakurai S, Oguni S, Hironaka M, Fukayama M, Morinaga S, Saito K. Mutations in c-kit gene exons 9 and 13 in gastrointestinal stromal tumors among Japanese. Jpn J Cancer Res 2001 May;92(5):494-498.
15. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of Experts. Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010 May;21(Suppl 5):v98-v102.
16. Gold JS, Gönen M, Gutiérrez A, Broto JM, García-del-Muro X, Smyrk TC, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localised primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol 2009 Nov;10(11):1045-1052.
|