
Colon Targeted Drug Delivery Systems: A Review on Primary and Novel Approaches
Anil K. Philip, Betty Philip
Philip AK, et al. OMJ. 25, 70-78 (2010); doi:10.5001/omj.2010.24
ABSTRACT
The colon is a site where both local and systemic delivery of drugs can take place. Local delivery allows topical treatment of inflammatory bowel disease. However, treatment can be made effective if the drugs can be targeted directly into the colon, thereby reducing the systemic side effects. This review, mainly compares the primary approaches for CDDS (Colon Specific Drug Delivery) namely prodrugs, pH and time dependent systems, and microbially triggered systems, which achieved limited success and had limitations as compared with newer CDDS namely pressure controlled colonic delivery capsules, CODESTM, and osmotic controlled drug delivery which are unique in terms of achieving in vivo site specificity, and feasibility of manufacturing process.
Department of Pharmaceutics, School of Pharmacy, University of Nizwa, Birkat Al Mouz, Nizwa-616, Sultanate of Oman.
Received: 07 Feb 2010
Accepted: 14 Mar 2010
Address correspondence and reprint request to: Dr. Anil K. Philip, Assistant Professor Department of Pharmaceutics, School of Pharmacy, College of Pharmacy and Nursing, University of Nizwa, Birkat Al Mouz, Nizwa-616, Sultanate of Oman.
E-mail: philip@unizwa.edu.om, philipanil23@yahoo.co.in
Philip AK, et al. OMJ. 25, 70-78 (2010); doi:10.5001/omj.2010.24
INTRODUCTION
Targeted drug delivery into the colon is highly desirable for local treatment of a variety of bowel diseases such as ulcerative colitis, Crohn’s disease, amebiosis, colonic cancer, local treatment of colonic pathologies, and systemic delivery of protein and peptide drugs.1,2 The colon specific drug delivery system (CDDS) should be capable of protecting the drug en route to the colon i.e. drug release and absorption should not occur in the stomach as well as the small intestine, and neither the bioactive agent should be degraded in either of the dissolution sites but only released and absorbed once the system reaches the colon.3 The colon is believed to be a suitable absorption site for peptides and protein drugs for the following reasons; (i) less diversity, and intensity of digestive enzymes, (ii) comparative proteolytic activity of colon mucosa is much less than that observed in the small intestine, thus CDDS protects peptide drugs from hydrolysis, and enzymatic degradation in duodenum and jejunum, and eventually releases the drug into ileum or colon which leads to greater systemic bioavailability.4 And finally, because the colon has a long residence time which is up to 5 days and is highly responsive to absorption enhancers.5
Oral route is the most convenient and preferred route but other routes for CDDS may be used. Rectal administration offers the shortest route for targeting drugs to the colon. However, reaching the proximal part of colon via rectal administration is difficult. Rectal administration can also be uncomfortable for patients and compliance may be less than optimal.6 Drug preparation for intrarectal administration is supplied as solutions, foam, and suppositories. The intrarectal route is used both as a means of systemic dosing and for the delivery of topically active drug to the large intestine. Corticosteroids such as hydrocortisone and prednisolone are administered via the rectum for the treatment of ulcerative colitis. Although these drugs are absorbed from the large bowel, it is generally believed that their efficacy is due mainly to the topical application. The concentration of drug reaching the colon depends on formulation factors, the extent of retrograde spreading and the retention time. Foam and suppositories have been shown to be retained mainly in the rectum and sigmoid colon while enema solutions have a great spreading capacity.7
Because of the high water absorption capacity of the colon, the colonic contents are considerably viscous and their mixing is not efficient, thus availability of most drugs to the absorptive membrane is low. The human colon has over 400 distinct species of bacteria as resident flora, a possible population of up to 1010 bacteria per gram of colonic contents. Among the reactions carried out by these gut flora are azoreduction and enzymatic cleavage i.e. glycosides.8 These metabolic processes may be responsible for the metabolism of many drugs and may also be applied to colon-targeted delivery of peptide based macromolecules such as insulin by oral administration.
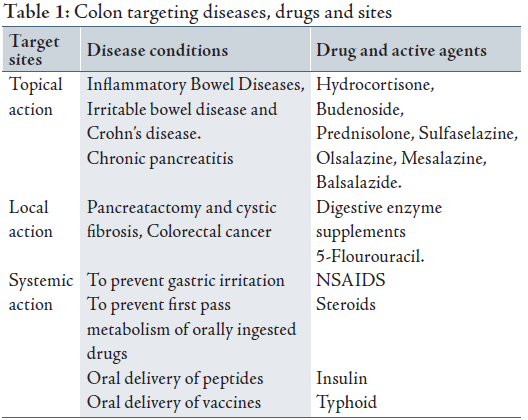
Target sites, colonic disease conditions, and drugs used for treatment are shown in Table 1.9
Advantages of CDDS over Conventional Drug Delivery
Chronic colitis, namely ulcerative colitis, and Crohn’s disease are currently treated with glucocorticoids, and other anti-inflammatory agents.10 Administration of glucocorticoids namely dexamethasone and methyl prednisolone by oral and intravenous routes produce systemic side effects including adenosuppression, immunosuppression, cushinoid symptoms, and bone resorption.11 Thus selective delivery of drugs to the colon could not only lower the required dose but also reduce the systemic side effects caused by high doses.12
Criteria for Selection of Drug for CDDS
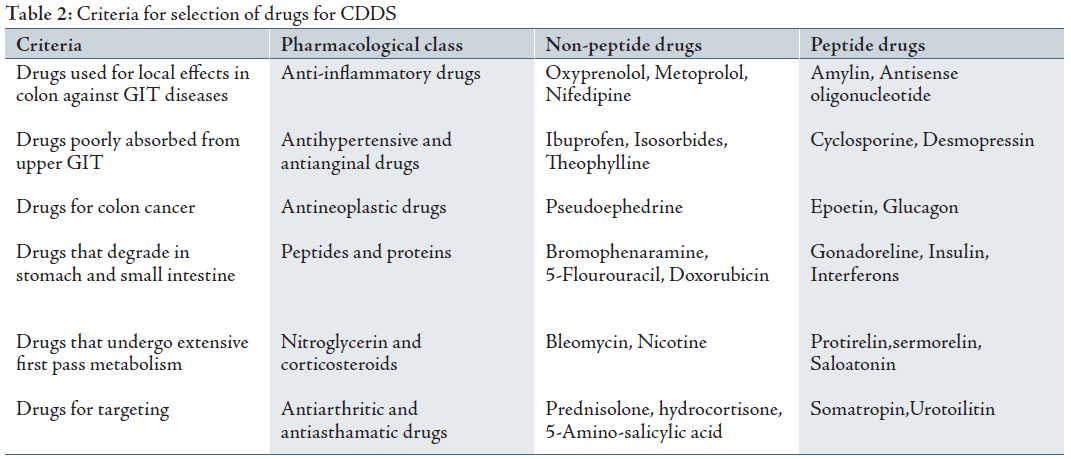
The best Candidates for CDDS are drugs which show poor absorption from the stomach or intestine including peptides. The drugs used in the treatment of IBD, ulcerative colitis, diarrhea, and colon cancer are ideal candidates for local colon delivery.13 The criteria for selection of drugs for CDDS is summarized in Table 2.14-16
Drug Carrier is another factor which influences CDDS. The selection of
carrier for particular drugs depends on the physiochemical nature of the drug as
well as the disease for which the system is to be used. Factors such as chemical
nature, stability and partition coefficient of the drug and type of absorption
enhancer chosen influence the carrier selection. Moreover, the choice of drug
carrier depends on the functional groups of the drug molecule.17 For
example, aniline or nitro groups on a drug may be used to link it to another
benzene group through an azo bond. The carriers, which contain additives like
polymers (may be used as matrices and hydro gels or coating agents) may
influence the release properties and efficacy of the systems.13
Approaches used for Site Specific Drug Delivery to Colon (CDDS)
Several approaches are used for site-specific drug delivery. Among the primary approaches for CDDS, These include:
1) Primary Approaches for CDDS
a. pH Sensitive Polymer Coated Drug Delivery to the Colon
In the stomach, pH ranges between 1 and 2 during fasting but increases after eating.21 The pH is about 6.5 in the proximal small intestine, and about 7.5 in the distal small intestine.22 From the ileum to the colon, pH declines significantly. It is about 6.4 in the cecum. However, pH values as low as 5.7 have been measured in the ascending colon in healthy volunteers.23 The pH in the transverse colon is 6.6 and 7.0 in the descending colon. Use of pH dependent polymers is based on these differences in pH levels. The polymers described as pH dependent in colon specific drug delivery are insoluble at low pH levels but become increasingly soluble as pH rises.24 Although a pH dependent polymer can protect a formulation in the stomach, and proximal small intestine, it may start to dissolve in the lower small intestine, and the site-specificity of formulations can be poor.25 The decline in pH from the end of the small intestine to the colon can also result in problems, lengthy lag times at the ileo-cecal junction or rapid transit through the ascending colon which can also result in poor site-specificity of enteric-coated single-unit formulations.24
b. Delayed (Time Controlled Release System) Release Drug Delivery to ColonTime controlled release system (TCRS) such as sustained or delayed release dosage forms are also very promising drug release systems. However, due to potentially large variations of gastric emptying time of dosage forms in humans, in these approaches, colon arrival time of dosage forms cannot be accurately predicted, resulting in poor colonical availability.26 The dosage forms may also be applicable as colon targeting dosage forms by prolonging the lag time of about 5 to 6 h. However, the disadvantages of this system are:
i. Gastric emptying time varies markedly between subjects or in a manner dependent on type and amount of food intake.
ii. Gastrointestinal
movement, especially peristalsis or contraction in the stomach would result in
change in gastrointestinal transit of the drug.27
iii. Accelerated transit through different regions of the colon has been observed in patients with the IBD, the carcinoid syndrome and diarrhea, and the ulcerative colitis.9, 28,29
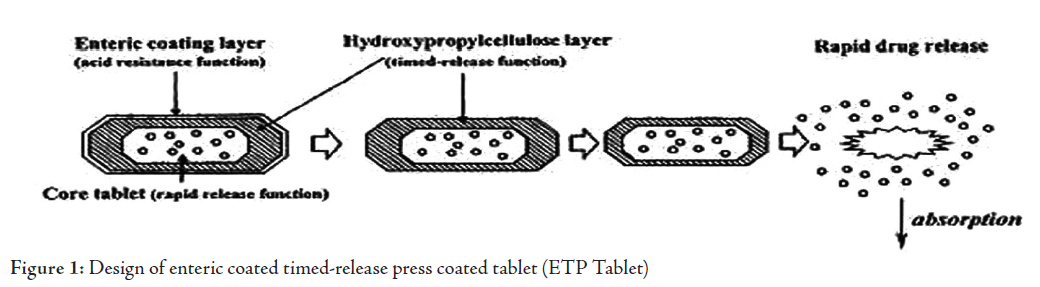
Therefore, time dependent systems are not ideal to deliver drugs to the colon specifically for the treatment of colon related diseases. Appropriate integration of pH sensitive and time release functions into a single dosage form may improve the site specificity of drug delivery to the colon. Since the transit time of dosage forms in the small intestine is less variable i.e. about 3±1 hr.30 The time-release function (or timer function) should work more efficiently in the small intestine as compared the stomach. In the small intestine drug carrier will be delivered to the target side, and drug release will begin at a predetermined time point after gastric emptying. On the other hand, in the stomach, the drug release should be suppressed by a pH sensing function (acid resistance) in the dosage form, which would reduce variation in gastric residence time.27 Enteric coated time-release press coated (ETP) tablets, are composed of three components, a drug containing core tablet (rapid release function), the press coated swellable hydrophobic polymer layer (Hydroxy propyl cellulose layer (HPC), time release function) and an enteric coating layer (acid resistance function).26,31 The tablet does not release the drug in the stomach due to the acid resistance of the outer enteric coating layer. After gastric emptying, the enteric coating layer rapidly dissolves and the intestinal fluid begins to slowly erode the press coated polymer (HPC) layer. When the erosion front reaches the core tablet, rapid drug release occurs since the erosion process takes a long time as there is no drug release period (lag phase) after gastric emptying. The duration of lag phase is controlled either by the weight or composition of the polymer (HPC) layer. (Fig. 1)
c. Microbially Triggered Drug Delivery to Colon
The microflora of the colon is in the range of 1011 -1012 CFU/mL, consisting mainly of anaerobic bacteria, e.g. bacteroides, bifidobacteria, eubacteria, clostridia, enterococci, enterobacteria and ruminococcus etc.28 This vast microflora fulfills its energy needs by fermenting various types of substrates that have been left undigested in the small intestine, e.g. di- and tri-saccharides, polysaccharides etc.32,33 For this fermentation, the microflora produces a vast number of enzymes like glucoronidase, xylosidase, arabinosidase, galactosidase, nitroreductase, azareducatase, deaminase, and urea dehydroxylase.34 Because of the presence of the biodegradable enzymes only in the colon, the use of biodegradable polymers for colon-specific drug delivery seems to be a more site-specific approach as compared to other approaches.5 These polymers shield the drug from the environments of stomach and small intestine, and are able to deliver the drug to the colon. On reaching the colon, they undergo assimilation by micro-organism, or degradation by enzyme or break down of the polymer back bone leading to a subsequent reduction in their molecular weight and thereby loss of mechanical strength.35,36,37,38,39 They are then unable to hold the drug entity any longer.40
i) Prodrug Approach for Drug Delivery to Colon
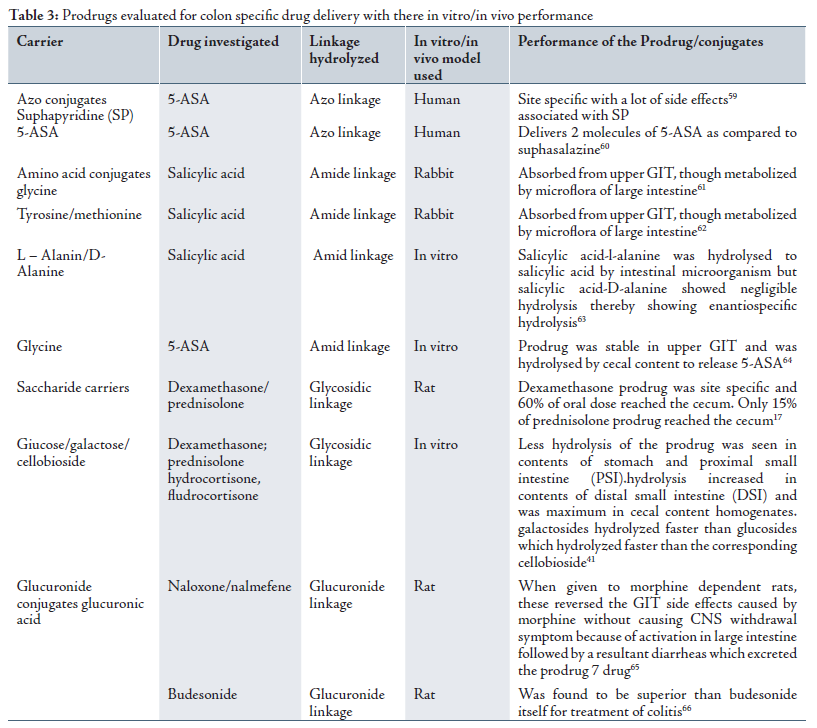
Prodrug is a pharmacologically inactive derivative of a parent drug molecule that requires spontaneous or enzymatic transformation in vivo to release the active drug. For colonic delivery, the prodrug is designed to undergo minimal hydrolysis in the upper tracts of GIT, and undergo enzymatic hydrolysis in the colon there by releasing the active drug moiety from the drug carrier. Metabolism of azo compounds by intestinal bacteria is one of the most extensively studied bacterial metabolic process.41 A number of other linkages susceptible to bacterial hydrolysis specially in the colon have been prepared where the drug is attached to hydrophobic moieties like amino acids, glucoronic acids, glucose, glactose, cellulose etc. Limitations of the prodrug approach is that it is not a very versatile approach as its formulation depends upon the functional group available on the drug moiety for chemical linkage. Furthermore, prodrugs are new chemical entities, and need a lot of evaluation before being used as carriers.42 A number of prodrugs have been outlined in Table 3.
(ii) Azo-Polymeric Prodrugs
Newer approaches are aimed at the use of polymers as drug carriers for drug delivery to the colon. Both synthetic as well as naturally occurring polymers have been used for this purpose. Sub synthetic polymers have been used to form polymeric prodrug with azo linkage between the polymer and drug moiety.18 These have been evaluated for CDDS. Various azo polymers have also been evaluated as coating materials over drug cores. These have been found to be similarly susceptible to cleavage by the azoreducatase in the large bowel. Coating of peptide capsules with polymers cross linked with azoaromatic group have been found to protect the drug from digestion in the stomach and small intestine. In the colon, the azo bonds are reduced, and the drug is released.31 A number of azo-polymeric prodrugs are outlined in Table 4.

iii) Polysaccharide Based Delivery Systems
The use of naturally occurring polysaccharides is attracting a lot of attention for drug targeting the colon since these polymers of monosaccharides are found in abundance, have wide availability are inexpensive and are available in a verity of a structures with varied properties. They can be easily modified chemically, biochemically, and are highly stable, safe, nontoxic, hydrophilic and gel forming and in addition, are biodegradable. These include naturally occurring polysaccharides obtained from plant (guar gum, inulin), animal (chitosan, chondrotin sulphate), algal (alginates) or microbial (dextran) origin. The polysaccrides can be broken down by the colonic microflora to simple saccharides.24 Therefore, they fall into the category of “generally regarded as safe” (GRAS). A number of polysaccharide-based delivery systems have been outlined in Table 5.
2. Newly Developed Approaches for CDDS
a. Pressure Controlled Drug-Delivery Systems
As a result of peristalsis, higher pressures are encountered in the colon than in the small intestine. Takaya et al. developed pressure controlled colon-delivery capsules prepared using ethylcellulose, which is insoluble in water.43 In such systems, drug release occurs following the disintegration of a water-insoluble polymer capsule because of pressure in the lumen of the colon. The thickness of the ethylcellulose membrane is the most important factor for the disintegration of the formulation.44,45 The system also appeared to depend on capsule size and density. Because of reabsorption of water from the colon, the viscosity of luminal content is higher in the colon than in the small intestine. It has therefore been concluded that drug dissolution in the colon could present a problem in relation to colon-specific oral drug delivery systems. In pressure controlled ethylcellulose single unit capsules the drug is in a liquid.46 Lag times of three to five hours in relation to drug absorption were noted when pressure-controlled capsules were administered to humans.
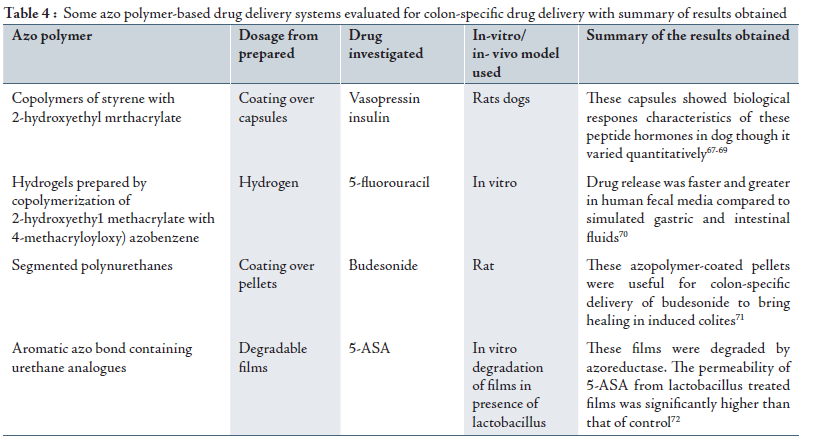
b. Novel Colon Targeted Delivery System (CODESTM)
CODESTM is an unique CDDS technology that was designed to avoid the inherent problems associated with pH or time dependent systems.47,48 CODESTM is a combined approach of pH dependent and microbially triggered CDDS. It has been developed by utilizing a unique mechanism involving lactulose, which acts as a trigger for site specific drug release in the colon, (Fig. 2). The system consists of a traditional tablet core containing lactulose, which is over coated with and acid soluble material, Eudragit E, and then subsequently overcoated with an enteric material, Eudragit L. The premise of the technology is that the enteric coating protects the tablet while it is located in the stomach and then dissolves quickly following gastric emptying. The acid soluble material coating then protects the preparation as it passes through the alkaline pH of the small intestine.49 Once the tablet arrives in the colon, the bacteria enzymetically degrade the polysaccharide (lactulose) into organic acid. This lowers the pH surrounding the system sufficient to effect the dissolution of the acid soluble coating and subsequent drug release.20
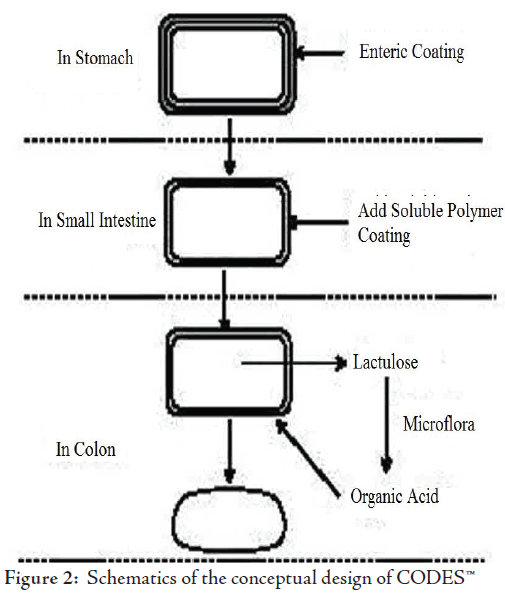
c. Osmotic Controlled Drug Delivery (ORDS-CT)
The OROS-CT (Alza corporation) can be used to target the drug locally to the colon for the treatment of disease or to achieve systemic absorption that is otherwise unattainable.50 The OROS-CT system can be a single osmotic unit or may incorporate as many as 5-6 push-pull units, each 4 mm in diameter, encapsulated within a hard gelatin capsule, (Fig. 3).51 Each bilayer push pull unit contains an osmotic push layer and a drug layer, both surrounded by a semipermeable membrane. An orifice is drilled through the membrane next to the drug layer. Immediately after the OROS-CT is swallowed, the gelatin capsule containing the push-pull units dissolves. Because of its drug-impermeable enteric coating, each push-pull unit is prevented from absorbing water in the acidic aqueous environment of the stomach, and hence no drug is delivered. As the unit enters the small intestine, the coating dissolves in this higher pH environment (pH >7), water enters the unit, causing the osmotic push compartment to swell, and concomitantly creates a flowable gel in the drug compartment. Swelling of the osmotic push compartment forces drug gel out of the orifice at a rate precisely controlled by the rate of water transport through the semipermeable membrane. For treating ulcerative colitis, each push pull unit is designed with a 3-4 h post gastric delay to prevent drug delivery in the small intestine. Drug release begins when the unit reaches the colon. OROS-CT units can maintain a constant release rate for up to 24 hours in the colon or can deliver drug over a period as short as four hours. Recently, new phase transited systems have come which promise to be a good tool for targeting drugs to the colon.52-55 Various in vitro / in vivo evaluation techniques have been developed and proposed to test the performance and stability of CDDS.
For in vitro evaluation, not any standardized evaluation technique is available for evaluation of CDDS because an ideal in vitro model should posses the in-vivo conditions of GIT such as pH, volume, stirring, bacteria, enzymes, enzyme activity, and other components of food. Generally, these conditions are influenced by the diet, physical stress, and these factors make it difficult to design a slandered in-vitro model. In vitro models used for CDDS are:
a) In vitro dissolution test
Dissolution of controlled-release formulations used for colon-specific drug delivery are usually complex, and the dissolution methods described in the USP cannot fully mimic in vivo conditions such as those relating to pH, bacterial environment and mixing forces.20 Dissolution tests relating to CDDS may be carried out using the conventional basket method. Parallel dissolution studies in different buffers may be undertaken to characterize the behavior of formulations at different pH levels. Dissolution tests of a colon-specific formulation in various media simulating pH conditions and times likely to be encountered at various locations in the gastrointestinal tract have been studied.56 The media chosen were, for example, pH 1.2 to simulate gastric fluid, pH 6.8 to simulate the jejunal region of the small intestine, and pH 7.2 to simulate the ileum segment. Enteric-coated capsules for CDDS have been investigated in a gradient dissolution study in three buffers. The capsules were tested for two hours at pH 1.2, then one hour at pH 6.8, and finally at pH 7.4.57
b) In vitro enzymatic tests
Incubate carrier drug system in fermenter containing suitable medium for bacteria (strectococcus faccium and B. Ovatus). The amount of drug released at different time intervals are determined. Drug release study is done in buffer medium containing enzymes (ezypectinase, dextranase), or rat or guinea pig or rabbit cecal contents. The amount of drug released in a particular time is determined, which is directly proportional to the rate of degradation of polymer carrier.
c) In vivo evaluation
A number of animals such as dogs, guinea pigs, rats, and pigs are used to evaluate the delivery of drug to colon because they resemble the anatomic and physiological conditions as well as the microflora of human GIT. While choosing a model for testing a CDDS, relative model for the colonic diseases should also be considered. Guinea pigs are commonly used for experimental IBD model. The distribution of azoreductase and glucouronidase activity in the GIT of rat and rabbit is fairly comparable to that in the human.58 For rapid evaluation of CDDS, a novel model has been proposed. In this model, the human fetal bowel is transplanted into a subcutaneous tullel on the back of thymic nude mice, which bascularizes within four weeks, matures, and becomes capable of developing of mucosal immune system from the host.
Drug Delivery Index (DDI) and Clinical Evaluation of Colon-Specific Drug Delivery Systems
DDI is a calculated pharmacokinetic parameter, following single or multiple dose of oral colonic prodrugs. DDI is the relative ratio of RCE (Relative colonic tissue exposure to the drug) to RSC (Relative amount of drug in blood i.e. that is relative systemic exposal to the drug). High drug DDI value indicates better colon drug delivery. Absorption of drugs from the colon is monitored by colonoscopy and intubation. Currently, gamma scintigraphy and high frequency capsules are the most preferred techniques employed to evaluate colon drug delivery systems.
CONCLUSION
The colonic region of the GIT has become an increasingly important site for drug delivery and absorption. CDDS offers considerable therapeutic benefits to patients in terms of both local and systemic treatment. Colon specificity is more likely to be achieved with systems that utilize natural materials that are degraded by colonic bacterial enzymes. Considering the sophistication of colon-specific drug delivery systems, and the uncertainty of current dissolution methods in establishing possible in-vitro/in-vivo correlation, challenges remain for pharmaceutical scientists to develop and validate a dissolution method that incorporates the physiological features of the colon, and yet can be used routinely in an industry setting for the evaluation of CDDS.
The authors reported no conflict of interest. The authors alone are responsible for the content and writing of the paper and no funding has been received on this work.
-
Philip AK, Dabas S, Pathak K. Optimized prodrug approach: A means for achieving enhanced anti-inflammatory potential in experimentally induced colitis. J Drug Target 2009; 17:235-241.
-
Oluwatoyin AO, John TF. In vitro evaluation of khaya and albizia gums as compression coating for drug targeting to the colon. J Pharm Pharmacol 2005; 57: 63-168.
-
Akala EO, Elekwachi O, Chase V, Johnson H, Marjorie L, Scott K. Organic redox initiated polymerization process for the fabrication of hydrogel for colon specific drug delivery. Drug Dev Ind Pharm 2003; 29:375-386.
-
Chourasia MK, Jain S K. Pharmaceutical approaches to colon targeted drug delivery systems. J Pharm Sci 2003; 6:33-66.
-
Basit A, Bloor J. Prespectives on colonic drug delivery, Business briefing. Pharmtech 2003; 185-190.
-
Watts P, Illum L. Colonic drug delivery. Drug Dev Ind Pharm 1997; 23:893-913.
-
Wood E, Wilson CG, Hardy JG. The spreading of foam and solution enemas. Int J Pharm 1985; 25:191-197.
-
Chien YW. Oral drug delivery and delivery systems. In: Chien YW, editor. Novel drug delivery systems. New York: Marcel Dekker Inc; 1992; 139-196.
-
Reddy MS, Sinha RV, Reddy DS. Colon targeted systems. Drugs Today 1999; 35(7):537.
-
Philip AK, Dubey RK, Pathak K. Optimizing delivery of flurbiprofen to the colon using a targeted prodrug approach. J Pharm Pharmacol 2008; 60:607-613.
-
Kulkarni SK. Pharmacology of gastro-intestinal tract (GIT). In: Kulkarni SK. editor, Handbook of experimental pharmacology. New Delhi: Vallabh Prakashan; 1999; 148-150.
-
McLeod AD, Friend DR, Thoma NT. Glucocorticoid-dextran conjugates as potential prodrugs for colon specific delivery- hydrolysis in rat gastrointestinal tract contents. J Pharm Sci 1994; 83(9):1284-1288.
-
Vyas SP, Khar RK. Gastroretentive systems. In: Vyas SP, Khar RK, editors. Controlled drug delivery: concepts and advances. New Delhi: Vallabh Prakashan, 2005; 218-253.
-
Antonin KH, Rak R, Bieck PR, Preiss R, Schenker U, Hastewell J, Fox R, Mackay M, The absorption of human calcitonin from the transverse colon of man. Int J Pharm 1996; 130(1):33-39.
-
Fara JW. Novel Drug Delivery and its Therapeutic Application. In: Presscot LF, Nimmo WS, editors. Colonic drug absorption and metabolism. Wiley: Chichester, 1989; 103-120.
-
Mackay M, Tomlinson E. Colonic delivery of therapeutic peptides and proteins, In: Biek PR, editors. Colonic drug absorption and metabolism. New York: Marcel Dekker, 1993; 159-176.
-
Friend DR, Chang GW. A colon-specific drug delivery system based on drug glycosides and glycosidase of colonic bacteria. J Med Chem 1984; 27: 261-266.
-
Mooter GV, Samyn C, Kinget R. In vitro evaluation of a colon specific drug delivery system: An absorption study of theophylline from capsules coated with azo polymers in rats. Pharm Res 1995; 12(2):244-247.
-
19. Prasad RYV, Krishnaiah YSR, Satyanarayana S. Trends in colonic drug delivery: A review. Ind Drugs 1996; 33:1-10.
-
Yang L, James S, Joseph A. Colon specific drug delivery new approaches and in vitro/ in vivo evaluation. Int J Pharm 2002; 235:1 -15.
-
Rubinstein A. Approaches and opportunities in colon-specific drug delivery. Crit. Rev Ther. Drug carrier Syst 1995; 12:101-149.
-
Evans DF, Pye G, Bramley R, Clark AG, Dyson TJ, Hardcastle JD. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 1988; 29:1035-1041.
-
Bussemer T, Otto I, Bodmeier R. Pulsatile drug-delivery systems. Crit Rev Ther Drug Carr Sys 2001; 18:433-458.
-
Ashord M, Fell JT, Attwood D, Sharma H, Woodhead P. An evaluation of pectin as a carrier for drug targeting to the colon. J Control Rel 1993; 26:213-220.
-
Fukui E, Miyamura N, Kobayashi M. An in vitro investigation of the suitability of presscoated tablets with hydroxypropylmethylcellulose acetate succinate (HPMCAS) and hydrophobicn additives in the outer shell for colon targeting. J Control Rel 200; 70:97-107.
-
Gazzaniga A, Iamartino P, Maffino G, Sangalli ME. Oral delayed release system for colonic specific drug delivery. Int J Pharm 1994; 108:77-83.
-
Fukui E, Miyamura N, Verma K, Kobayashi M. Preparation of enteric coated time released press coated tablets and evaluation of their function by in vitro and in vivo tests for colon targeting. Int J Pharm 2000; 204:7-15.
-
Vassallo M, Camilleri M, Phillip SF, Brow ML, Chapman NJ, Thomforde GM. Transit through the proximal colon influences stool weight in the a irritable bowel syndrome. Gastroenterology 1992; 102:102-108.
-
Vonderohe MR, Camolleri M, Kvols LK, Thomforde GM. Motor dysfunction of the small bowel and colon in patients with the carcinoid syndrome and diarrhea. New Eng J Med 1993; 329:1073-1078.
-
Kinget R, Kalala W, Vervoort L, Mooter G. Colonic drug delivery. J Drug Target 1998; 6:129-149.
-
Hita V, Singh R, Jain SK. Colonic targeting of metronidazole using azo aromatic polymers, development and characterization. Drug Del 1997; 4:19-22.
-
Rubunstein A. Microbially controlled drug delivery to the colon. Biopharm Drug Dispos 1990; 11:465-475.
-
Cummings JH, Englyst HN. (1987) Fermentation in the human large intestine and available substrates. Am J Clin Nutri 1987; 45:1243-1255.
-
Scheline RR. Metabolism of foreign compounds by gastrointestinal microorganisms. Pharmacol Rev 1973; 25:451-523.
-
Peters R, Kinget R. Film-forming polymers for colonic drug deliver: Synthesis and physical and chemical properties of methyl derivatives of Eudragit S. Int J Pharm 1993; 94:125-134.
-
Huang SI, Bansleben DA, Knox JR. Biodegradable polymers: Chymotrypsin degradation of low molecular weight poly (ester-urea) containing phenylalanine. J App Poly Sci 1979; 23:429-437.
-
Swift G. Biodegradable polymers in the environment: are they really biodegradable. Proc ACS Div Poly Mat Sci Eng 1992; 66:403-404.
-
Ratner BD, Gladhill KW, Horbett TA. Analysis of in vitro enzymatic and oxidative degradation of polyurethanes. J Biomed Mat Res 1988; 22:509-527.
-
Hergenrother RW, Wabewr HD, Cooper SL. The effect of chain extenders and stabilizers on the in vivo stability of polyurethanes. J App Biomat 1992; 3:17-22.
-
Park K, Shalaby WSW, Park H. editors, Biodegradation In: Biodegradable hydrogels for drug delivery, USA: Technomic publishing company, 1993; 13-34.
-
Friend DR, Chang GW. Drug Glycosides: Potential prodrugs for colon specific drug delivery. J Med Chem 1985; 28:51-57.
-
Sinha VR, Kumria R. Microbially triggered drug delivery to the colon. Eur J Pharm Sci 2003; 18:3-18.
-
Takaya T, Niwa K, Muraoka M, Ogita I, Nagai N, Yano R, Kimura G, Yoshikawa Y, Yoshikawa H, Takada K. Importance of dissolution process on systemic availability of drugs delivered by colon delivery system. J Control Rel 1998; 50 (1-3):111-122.
-
Muraoka M, Hu Z, Shimokawa T, Sekino S, Kurogoshi R, Kuboi Y, Yoshikawa Y, Takada K. Evaluation of intestinal pressure-controlled colon delivery capsule containing caffeine as a model drug in human volunteers. J Control Rel 1998; 52(1-2):119-129.
-
Jeong Y, Ohno T, Hu Z, Yoshikawa Y, Shibata N, Nagata S, Takada K. Evaluation of an intestinal pressure-controlled colon delivery capsules prepared by a dipping method. J Control Rel 71(2):175-182.
-
Hay DJ, Sharma H, Irving MH. Spread of steroid containing foam after intrarectal administration. Brit Med J 1979; 1:1751-1753.
-
Watanabe S, Kawai H, Katsuma M, Fukui M. Colon specific drug release system. U. S. Patent, 1998, 09/183339.
-
Takemura S, Watanabe S, Katsuma M, Fukui M. Human gastrointestinal treatment study of a novel colon delivery system (CODES) using scintography, Pro Int Sym Control Rel Bioact Mat 2000, 27.
-
Masataka K, Watanabe S, Takemura S, Sako K, Sawada T, Masuda Y, Nakamura K, Fukui M, Connor AL, Wilding IR. Scintigraphic evaluation of a novel colon-targeted delivery system (CODESTM) in healthy volunteers. J Pharm Sci 2004; 93(5):1287-1299.
-
Theeuwes F, Guittared G, Wong P. Delivery of drugs to colon by oral dosage forms. U. S. Patent, 4904474
-
Swanson D, Barclay B, Wong P, Theeuwes F. Nifedipine gastrointestinal therapeutics system. Am J Med 1987; 8(6):3.
-
Philip AK, Pathak K. Osmotic flow through asymmetric membrane: A means for controlled delivery of drugs with varying solubility. AAPS PharmSciTech 2006; 7(3):1-11.
-
Philip AK, Pathak K. In situ-formed asymmetric membrane capsule for osmotic release of poorly water-soluble drug. PDA J Pharm Sci Tech 2007; 61(1):24-36.
-
Philip AK, Pathak K, Shakya P. Asymmetric membrane in membrane capsules: A means for achieving delayed and osmotic flow of cefadroxil. Eur J Pharm Biopharm 2008; 69(2):658-666.
-
Philip AK, Pathak K. Wet process induced phase transited drug delivery system: A means for achieving osmotic, controlled, and level a ivivc for poorly water soluble drug. Drug Dev Ind Pharm 2008; 34(7):735-743.
-
Ahmed IS. Effect of simulated gastrointestinal condition on drug release from pectin/ethyl cellulose as film coating for drug delivery to the colon. Drug Dev Ind Pharm 2005; 31(4-5): 465-470.
-
Cole ET, Scott RA, Connor AL, Wilding IR, Petereit HU, Schminke C, Beckert T, Cadé D. Enteric coated HPMC capsules designed to achieve intestinal targeting. Int J Pharm 2002; 231(1):83-95.
-
Mooter VG, Kinget R. Oral colon-specific drug delivery: A review: Drug Delivery 1995; 2:881-931.
-
Khan AK, Piris J, Truelone SC. An experiment to determine the active therapeutic moiety of sulphasalazine. Lancet 1977; 2:895-896.
-
Chan RP, Pope DJ, Gilbett AP, Sneta PJ, Baron JH, Bennardjones JF. Studies of two novel sulphasalazine analogs ip salazide and balsalazide. Digest Diseases Sci 1983; 28:609-716.
-
Shibasaki J, Inoue Y, Kadoskai YK, Sasaki H, Nakamura J. Hydrolysis of salicyluric acid in rabbit intestinal microorganisms. J Pharmacobio-Dyn 1885; 8:989-995.
-
Nakamura J, Kido M, Nishida K, Sasaki H. Hydrolysis of salicylic acid tyrosine salicylic acid-methionine prodrug in rabbits. Int J Pharm 1992; 87: 59-66.
-
Nakamura J, Tagami C, Nishida K, Saskai H. Unequal hydrolysis of salicylic acid-D-alanine and salicylic acid-L-alanine conjugate in rabbit intestinal microorganisms. Chem Pharm Bull 1992b; 40(2):547-549.
-
Jung YJ, Lee JS, Kim HH, Kim YK, Han SK. Synthesis and evaluation of 5-aminosalicylicylglycine as a potential colon specific prodrug of 5-aminosalicylic acid. Arch Pharmacol Res 1998; 21:174-178.
-
Simpkins JW, Smulkowski M, Dixon R, Tuttle R. Evidence for the delivery of narcotic antagonists to the colon as their glucuro-nide conjugates. J Pharmacol Exp Thera 1988; 244:195-205.
-
Cui N, Friend DR, Fedora RN. A budesonide prodrug accelerates of colitis in rats. Gut 1994; 35:1439-1446.
-
Saffron M, Kumar GS, Savariora C, Burnham JC, Williams F, Neekers DC. A new approach to the oral administration of insulin and other peptide drugs. Sci 1986; 233:1081-1084.
-
Saffron M, Bedra C, Kumar GS, Neckers DC. Vasopressin: A model for the study of effects of additives on the oral and rectal administration of peptide drugs, J Pharm Sci 1988; 77:33-38.
-
Saffron M, Field JB, Pena J, Jones RH, Ohuda Y. Oral insulin in diabetic dogs, J Endocrinol 1991; 131:267-278.
-
Shanta KL, Ravichandran P, Rao KP. Azopolymeric hydrogels for colon targeted drug delivery. Biomat 1995; 16:1313-1318.
-
Tozaki H, Fujita T, Komoike J, Kim SI, Terashima H, Muranishi S, Okabe S, Yamamoto A. Colon-specific delivery of budesonide with azopolymer coated pellets: therapeutic effects of budesonnide with a novel dosage from against 2,4,6-trinitrobenzenesul-phonic acid-inducad colitis in rats. J Pharm Pharmacol 1999; 51:257-261.
-
Chavan MS, Sant VP, Nagarsenker MS. Azo-containing urethane analogues for colonic drug delivery: synthesis, characterization and in vitro evaluation. J Pharm Pharmacol 2001; 53:895-900.
-
Tozaki H, Komoike J, Tada C, Maruyama T, Terabe A, Suzuki T, Yamamoto A, Muranishi S. Chitosan capsules for colon-specific drug delivery: improvement of insulin absorption from the rat colon. J Pharm Sci 1997; 86: 1016-1021.
-
Aiedeh K, Taha MO. Synthesis of chitosan succinate and chitosan phthalate and their evaluation as suggested matrices in orally administered colon specific drug delivery system. Arch Pharmacol Res 1999; 332:103-107.
-
Rubinstein A, Radai R, Ezra M, Pathak S, Rokem JS. In vitro evaluation of calcium pectinate: A potential colon-specific drug delivery carrier. Pharm Res 1993; 10:258-263.
-
Wakerly Z, Fell J, Attwood D, Parkins D. Studies on amidated pectins as potential carriers in colonic drug delivery. J Pharm Pharmacol 1997; 49:622-625.
-
Ahrabi SF, Madseh G, Dyrstad K, Sande SA, Graffner C. Development of pectin matrix tablets for colonic delivery of model drug ropivacanie. Eur J Pharm Sci 2000; 10:43-52.
-
Rubinstin A, Nakar D, Sintov A. Chondroitin sulphate: A potential biodegradable carrier for colon-specific drug delivery Int J Pharm 1992; 84: 141-150.
-
Shun YL, Ayres JW. Calcium alginate beads as core carriers of 5-aminosalicylic acid. Pharm Res 1992; 9:714-790.