| |
Introduction
The autosomal-dominantly inherited 1.4Mb tandem duplication in the PMP22 gene at locus 17p11.2 most frequently manifests as hereditary demyelinating, sensorimotor polyneuropathy with weakness and wasting of the lower limbs, sensory disturbances, and foot deformity (HMSN-1A).1 Rare clinical features include sensorineural hearing-loss, moderate (motor) developmental delay, or gait disturbance.2 As disease severity may vary greatly; marked interfamilial and interfamilial phenotypic variability may be observed. Hypertelorism has rarely been reported.
Case Report
The patient was a 31 year-old Caucasian male with an uneventful history until adulthood, who then developed recurrent sensory disturbances, when taking certain positions, which resolved upon changing of the limb position, muscle cramps of the thighs, and tinnitus. Psychomotor development was normal and motor milestones were reached at time. Clinical neurologic examination revealed mild facial dysmorphism, manifesting as stuck out ears and hypertelorism (Fig. 1), with an interorbital distance of 41 mm (normal: 18-35 mm),3 an interpupillary distance of 86 mm, intention tremor, brady-diadochokinesia, reduced tendon reflexes, discrete weakness for foot extension and markedly reduced nerve conduction velocities, (Table 1). Cerebral MRI was normal. He was diagnosed as HMSN-1A after the disorder had been diagnosed before in his 27 year-old half-sister who presented with short stature (163 cm), sensorimotor polyneuropathy with distal quadruparesis, numbness of the distal limbs, tremor, ataxia, sensorineural hearing-loss, and hypothyroidism (Table 1). HMSN-1A was also diagnosed in the father of the two, who had developed sensorimotor deficits, gait disturbance, and foot deformity since age 30 years without other features as in his daughter or son, (Table 1). Psychomotor development and achievement of motor milestones were normal in both father and half-sister.
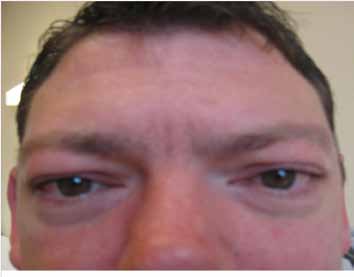
Figure 1: A 31 years-old Caucasian male carrying the common tandem duplication in the PMP22 gene locus showed hypertelorism and facial dysmorphism. He additionally presented with hereditary neuropathy (HMSN-1A) and liability to pressure palsies.
Table 1: Clinical and electrophysiological findings of the index patient, his half-sister and their father.
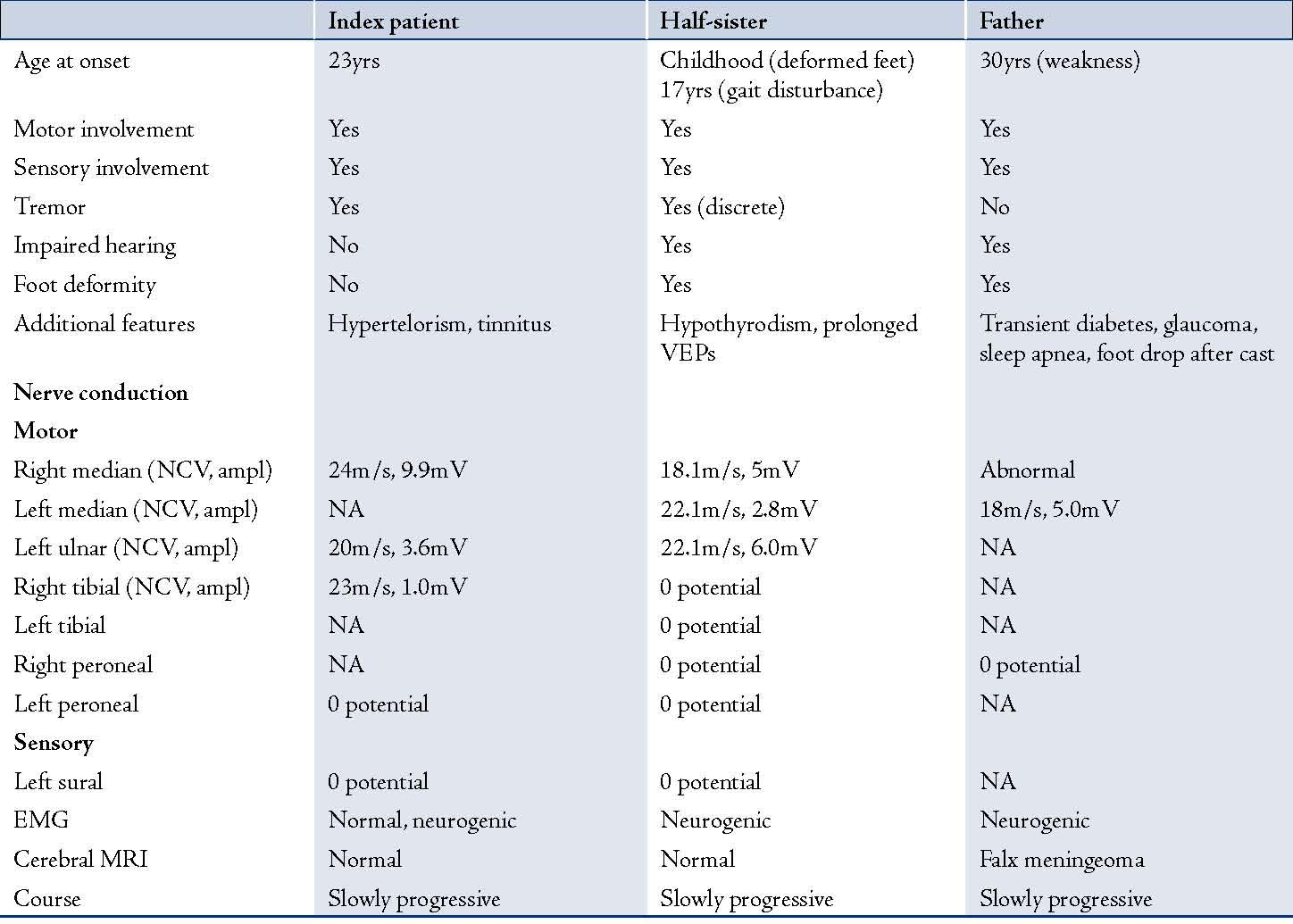
NCV: nerve conduction velocity, ampl: nerve action potential amplitude, EMG: electromyography, NA: not available, VEP: visually evoked potentia
The application of the microsatellite analysis according to Badano et al. 2001,4 using the markers D17S2220, D17S2224, D17S2226, D17S2227, and D17S2230 revealed the common 1.4 Mb tandem duplication in the PMP22 gene at 17p11.2 in the father as well as in his two children. No flanking markers outside the commonly duplicated region were analysed.
Discussion
Dysmorphic features have only rarely been reported in patients with hereditary neuropathy. In a 4 months-old patient with the Charcot-Marie-Tooth (CMT) duplication on chromosome 17, coronal synostosis and club feet were described.5 An 8 years-old girl carrying four submicroscopic interspersed 17p duplications presented with mental retardation, short stature, microcephaly, and mild dysmorphic features including mild hypertelorism.5 Also, a 6 years-old boy carrying the de novo duplication 17p11.2p12, different from the usual 1.4Mb duplication in CMT1A, presented with behavioral deficits, moderate developmental delay, gait disturbance, autism, and mild dysmorphic features, including mild hypertelorism.2 Facial dysmorphism has also been described in a subtype of CMT4 (autosomal-recessive HMSN), also known as congenital cataracts, facial dysmorphism, and neuropathy (CCFDN) syndrome,6 which predominantly occurs in the European gypsy population. The CCFDN-syndrome may additionally present with microcornea, moderate cognitive deficit, pyramidal signs, mild chorea, short stature, or hypogonadotropic hypogonadism.7-10 Facial dysmorphism was also reported in an 8 years-old boy with congenital insensitivity to pain and anhidrosis (CIPA, HSAN-IV). He additionally presented with severe swallowing disorder and a myogenic EMG.11 The boy carried the c.C2011T mutation in exon 15 of the NTRK1 gene.11 Hypotelorism and unusual skin folds and creases have recently been described as additional phenotypic features in hereditary neuralgic amyotrophy (HNA).12 Dysmorphic features and neuropathy can also be found in peripheral neuropathy associated with agenesis of the corpus callosum (ACCPN) syndrome.13 The ACCPN syndrome is a severe autosomal recessive sensorimotor neuropathy associated with mental retardation, dysmorphic features, and complete or partial agenesis of the corpus callosum.13 ACCPN is due to mutations in the SLC12A6 gene on chromosome 15q, which encodes the K+-Cl- transporter KCC3.13 Dysmorphic facial and digital features were also reported in a child with partial trisomy of the 17p12pter region and HMSN1.14 In a 7 year-old girl with CMT1A due to the duplication 17p10-p12 dysmorphic facial features, dislocation of hips, talipes, developmental delay, premature adrenarche, and deep palmar creases supplemented the phenotype.15
Hypertelorism in the presented patient could be a phenotypic manifestation of the PMP22 mutation but could also be explained by the involvement of contiguous genes that lie adjacent or distant to the PMP22 gene in the 17p11.2 region. In patients with the duplication dup(17)(p12+p11.2) mild cognitive impairment was associated with subtle dysmorphic features, including hypertelorism.16,17 There has also been a report of a patient with mosaic trisomy 17 who presented with developmental delay and hypertelorism.18 Hypertelorism in the presented patient could also be the manifestation of a collateral mutation in a gene outside the duplicated region inherited from the mother and distinct from PMP22. It is also conceivable that the presented family did not carry the common 1.4Mb duplication, which usually has the same size, but one that was larger in size. Due to the limitations of the applied method, the exact boundaries of the duplication were not determined. Additionally, it can be speculated that the family carried not only the 17p11.2 duplication but also an undetected 17p12.2 deletion, which may phenotypically manifest as Smith-Magenis syndrome. In single cases of Smith-Magenis syndrome, hypertelorism has been reported.19 Since hypertelorism is frequently seen in many other genetic disorders, such as deletion 4p, 4q, 9p, 11q, or 13q; the occurance of hypertelorism in the presented patient is rather interpreted as coincidential than as casual. Though a second trouble can be assumed resulting in the coincidental occurrence of two independent phenotypes, it cannot be definitively excluded that hypertelorism was simply due to the PMP22 duplication. Assuming that hypertelorism resulted from the 17p11.2 duplication, it might be explained by a variant longer than the common 1.4Mb duplicated region, which also affected genes involved in the formation of the facial shape. Since flanking markers were not analysed, the size of the duplication might be longer than that of the common duplication.
To explain the phenotypic variability within the presented family, it can be speculated that additional polymorphisms or modifier genes influenced the variable phenotype. The phenotypic variability between the described family and the previously reported cases may be due to the same causes as the intra-familial variability. A single major anomaly can be found in 1.3% and a minor anomaly in 2% of the new-born babies.20 However, this is of little concern since the frequency of major defects is not appreciably increased in this group. In both children, the onset of clinical manifestations was earlier than in their father and clinical manifestations were more severe; a phenomenon known as anticipation. Anticipation has particularly been reported in tri- and tetra-nucleotid expansion disorders, which were not diagnosed in the presented family, but appropriate analysis of the phenomenon usually requires large pedigrees. Anticipation has not been reported in hereditary neuropathies so far. Anticipation in the presented family and phenotypic heterogeneity between the family members suggests a weak phenotype-genotype correlation, which is usually closer in patients with other hereditary neuropathies.21
Conclusion
The presented cases show that the common duplication 17p11.2 may present with marked intra-familial phenotype variability and that mild facial dysmorphism with stuck out ears and hypertelorism may be a rare clinical feature of this mutation. However, hypertelorism could also be attributed to the involvement of contiguous genes or to mutations in genes distinct from PMP22.
Acknowledgements
The author reported no conflict of interest and no funding was reported on this work.
References
1. Thomas PK. Overview of Charcot-Marie-Tooth disease type 1A. Ann N Y Acad Sci 1999 Sep;883:1-5.
2. Moog U, Engelen JJ, Weber BW, Van Gelderen M, Steyaert J, Baas F, et al. Hereditary motor and sensory neuropathy (HMSN) IA, developmental delay and autism related disorder in a boy with duplication (17)(p11.2p12). Genet Couns 2004;15(1):73-80.
3. Bayani L, Sadeghi Tari A, Hamzeh-Doost K, Kasaii A. The bony interorbital distance and orbital measurements in the Iranian population: A CT study. Ir J Rad 2006;3:173-178.
4. Badano JL, Inoue K, Katsanis N, Lupski JR. New polymorphic short tandem repeats for PCR-based Charcot-Marie-Tooth disease type 1A duplication diagnosis. Clin Chem 2001 May;47(5):838-843.
5. Vissers LE, Stankiewicz P, Yatsenko SA, Crawford E, Creswick H, Proud VK, et al. Complex chromosome 17p rearrangements associated with low-copy repeats in two patients with congenital anomalies. Hum Genet 2007 Jul;121(6):697-709.
6. Claramunt R, Sevilla T, Lupo V, Cuesta A, Millán JM, Vílchez JJ, et al. The p.R1109X mutation in SH3TC2 gene is predominant in Spanish Gypsies with Charcot-Marie-Tooth disease type 4. Clin Genet 2007 Apr;71(4):343-349.
7. Navarro C, Teijeira S. Neuromuscular disorders in the Gypsy ethnic group. A short review. Acta Myol 2003 May;22(1):11-14.
8. Seeman P, Sisková D. Autosomal recessive ethnic diseases of Czech Gypsies. Cas Lek Cesk 2006;145(7):557-560, discussion 561.
9. Tournev I, Kalaydjieva L, Youl B, Ishpekova B, Guergueltcheva V, Kamenov O, et al. Congenital cataracts facial dysmorphism neuropathy syndrome, a novel complex genetic disease in Balkan Gypsies: clinical and electrophysiological observations. Ann Neurol 1999 Jun;45(6):742-750.
10. Kalaydjieva L. Congenital Cataracts, Facial Dysmorphism, and Neuropathy. 2010 Mar 02. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Available from http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=ccfdn
11. Raspall-Chaure M, Del Toro-Riera M, Gratacós M, Cuenca-León E, Ferrer I, Indo Y, et al. Congenital insensitivity to pain with anhidrosis associated with congenital myasthenic syndrome. Rev Neurol 2005 Aug;41(4):218-222.
12. Jeannet PY, Watts GD, Bird TD, Chance PF. Craniofacial and cutaneous findings expand the phenotype of hereditary neuralgic amyotrophy. Neurology 2001 Dec;57(11):1963-1968.
13. Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet 2002 Nov;32(3):384-392.
14. Vogt J, Hill S, Brueton L. Partial trisomy 17p12pter, associated with pre and postnatal growth retardation, dysmorphic facial and digital features, developmental delay, and signs of HMSN1 in early childhood. Eur J Med Genet 2006 Sep-Oct;49(5):439-443.
15. Shaw CJ, Stankiewicz P, Christodoulou J, Smith E, Jones K, Lupski JR. A girl with duplication 17p10-p12 associated with a dicentric chromosome. Am J Med Genet A 2004 Jan;124A(2):173-178.
16. Magenis RE, Brown MG, Allen L, Reiss J. De novo partial duplication of 17p [dup(17)(p12----p11.2)]: clinical report. Am J Med Genet 1986 Jul;24(3):415-420.
17. Brown A, Phelan MC, Patil S, Crawford E, Rogers RC, Schwartz C. Two patients with duplication of 17p11.2: the reciprocal of the Smith-Magenis syndrome deletion? Am J Med Genet 1996 May;63(2):373-377.
18. Stankiewicz P, Parka SS, Holder SE, Waters CS, Palmer RW, Berend SA, et al. Trisomy 17p10-p12 resulting from a supernumerary marker chromosome derived from chromosome 17: molecular analysis and delineation of the phenotype. Clin Genet 2001 Nov;60(5):336-344.
19. Babovic-Vuksanovic D, Jalal SM, Garrity JA, Robertson DM, Lindor NM. Visual impairment due to macular disciform scars in a 20-year-old man with Smith-Magenis syndrome: another opthalmologic complication. Am J Med Genet 1998 Dec;80(4):373-376.
20. De Araújo AM, Salzano FM. Congenital malformations, twinning and associated variables in a Brazilian population. Acta Genet Med Gemellol (Roma) 1975;24(1-2):31-39.
21. Rotthier A, Baets J, De Vriendt E, Jacobs A, Auer-Grumbach M, Lévy N, et al. Genes for hereditary sensory and autonomic neuropathies: a genotype-phenotype correlation. Brain 2009 Oct;132(Pt 10):2699-2711.
|
|