
A Chronic Granulomatous Disease of Childhood
Reem Abdwani,¹ Rana Abdel Rahim,¹ Anuradha Ganesh,² Aysha Al-Hamdani³
OMJ JAN 2009 VOL.24 NO.1 P.56 doi:10.5001/omj.2009.16
A 5-year-old girl presented with a five-month history of low grade fever and arthralgia with decreased energy level. Examination revealed arthritis affecting both ankles and knees as well as hepatosplenomegaly. Funduscopic examination showed posterior granulomatous uveitis (Figure 1). Synovial biopsy revealed synovial proliferation with non-necrotizing granulomas (Figure 2).
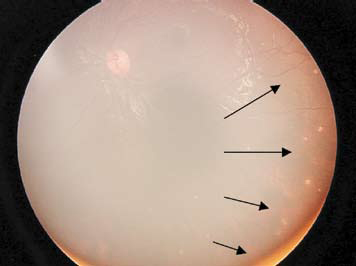
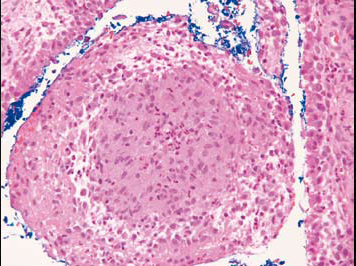
Figure 1:
Funduscopic images
Figure 2:
Microscopic synovial biopsy
Fundus – Multiple, discrete, hypo-pigmented choriodal lesions; some lesions are new (swollen, fuzzy margins) while some are old (punched-out).
Sections showed fragments of para-articular tissue lined by hyperplastic synoviumexhibiting papillary folds with congested blood vessels and moderate acute and chronic inflammatory cell infiltration consisting mainly of numerous plasma cells admixed with lymphocytes and occasional lymphoid follicles. Non necrotizing granulomas consisting of epithelioid cells were noted within the synovium. No acid fast bacilli seen and no fungal organisms noted on special stains .
QUESTION:
What is your diagnosis?
1- Systemic onset juvenile idiopathic arthritis
2- Tuberculosis
3- Histoplasmosis
4- Blau syndrome
5- Early onset sarcoidosis
ANSWER
All of the above answers should be included in the differential diagnosis when dealing with a child with granulomatous uveitis, arthritis and hepatosplenomegaly.
DISCUSSION
In a child presenting with granulomatous arthritis, uveitis and hepatosplenomegaly, some of the infectious organisms that need to be considered include mycobacteria and fungal causes such as coccidioidomycosis, histoplasmosis, and blastomycosis. Systemic onset Juvenile Idiopathic Arthritis (SoJIA) is another differential diagnosis to consider, which presents typically with fever and arthritis and can have systemic features including hepatosplenomegaly, lymphadenopathy, serositis or rash. However, posterior uveitis rarely complicates rheumatic diseases in children, and its presence suggests the diagnosis of sarcoidosis rather than juvenile idiopathic arthritis.
Blau syndrome and early onset sarcoidosis are now believed to belong to the same disease spectrum. Blau syndrome represents a familial autosomal dominant form while early onset sarcoidosis represents a sporadic form. They are characterized by inflammation of the skin, eye and joints, with a typical onset before five years. Patients exhibit a papular, erythematous rash, sometimes only transiently. Arthritis develops in the first decade of life with a remarkably proliferative synovitis described as ``boggy’’ or cyst-like. Preservation of range of motion and absence of erosive radiographic changes are characteristic. Pan-uveitis (less frequently anterior uveitis) is mostly bilateral and granulomatous. Secondary glaucoma is not uncommon and may lead to devastating visual loss. In addition to the classic triad, other atypical organ involvement including liver, peripheral nerves and kidneys 1-3 have been reported. In our patient, we suspected hepatic and splenic granulomas, a biopsy was not performed for confirmation. Both Blau syndrome and early onset sarcoidosis are associated with mutations in the gene encoding NOD2/CARD15 (located on chromosome 16; 16q12 4 diagnosis may be confirmed through skin, synovial or conjunctival biopsy, as well as by genetic testing. Histologically, there is synovial proliferation with non-caseating giant cell granulomas, as in our patient.
In our patient, all the work up was negative for infectious agents including Mantoux, synovial biopsy for AFB stain and culture. Although, granulomatous uveitis has been reported in patients with JIA,5 the presence of granuloma in the synovium is not seen in SoJIA excluding the diagnosis. Our clinical impression for this patient is Blau syndrome/ early onset sarcoidosis disease spectrum. Although the preliminary work up was negative,including angiotensin converting enzymes, serum and urinary calcium as well as hilar adenopathy on chest x-ray, we are still awaiting the confirmatory genetic testing.
DA. Granulomatous inflammation in juvenile idiopathic arthritis-associated uveitis. J AAPOS. 2008 Aug (abstract)REFERENCES
-
Ting SS, Ziegler J, Fischer E. Familial granulomatous arthritis (Blau syndrome) with granulomatous renal lesion. J Pediatr 1998; 133:450-452.
-
Saini SK, Rose CD. Liver involvement in familial granulomatous arthritis (Blau syndrome). J Rheumatol 1996; 23:396-399.
-
Jabs DA, Houk JL, Bids WB, Arnett FC. Familial granulomatous synovitis, uveitis, and cranial neuropathies. Am J Med 1985; 78:801-804.
-
Miceli-Richard C, Lesage S, Rybojad M, Prieur Am, Manouvrier-HAnu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nature Genet. 2001; 29:19-20.
-
Keenan JD, Tessler HH, Goldstein DA. Granulomatous inflammation in juvenile idiopathic arthritis-associated uveitis. J AAPOS. 2008 Aug(abstract)