
Caffey Disease or Infantile Cortical Hyperostosis: A Case Report
Narayanan Kutty, Doylene Thomas, Lionel George, Thomas B. John
Kutty N, et al. OMJ. 25, 134-136 (2010); doi:10.5001/omj.2010.36
ABSTRACT
Caffey disease or Infantile Cortical Hyperostosis (ICH) is a rare and mostly self limiting condition affecting young infants. It is characterized by acute inflammation of the periostium and the overlying soft tissue and is accompanied by systemic changes of irritability and fever. Diagnosis may be delayed as this disorder mimics a wide range of diseases including osteomyelitis, hypervitaminosis A, scurvy, bone tumors and child abuse. The emphasis here is to remind clinicians about the existence of the disease in this country.
From the Department of Child Health, Sultan Qaboos Hospital, Salalah, Sultanate of Oman
Received: 12 Dec 2009
Accepted:19 Jan 2010
Address correspondence and reprint request to: Dr. Narayanan Kutty, Department of Child Health, Sultan Qaboos hospital, Salalah, Sultanate of Oman
E-mail: nkbpanicker@gmail.com
Kutty N, et al. OMJ. 25, 134-136 (2010); doi:10.5001/omj.2010.36
INTRODUCTION
Caffey disease or Infantile Cortical Hyperostosis (ICH) is a rare and mostly self limiting condition affecting young iCaffey disease or Infantile Cortical Hyperostosis (ICH) is a rare and mostly self limiting condition affecting young infants. It is characterized by acute inflammation of the periostium and the overlying soft tissue and is accompanied by systemic changes of irritability and fever. Diagnosis may be delayed as this disorder mimics a wide range of diseases including osteomyelitis, hypervitaminosis A, scurvy, bone tumors and child abuse. The emphasis here is to remind clinicians about the existence of the disease in this country. While there are no laboratory tests to confirm the diagnosis of Caffey disease, a high index of suspicion in a typical clinical setting can avoid protracted investigations for this otherwise self-limiting illness.
A 4 month old Omani boy was admitted to Sultan Qaboos Hospital, Salalah, with a three week history of intermittent low grade fever, excessive crying and irritability. There were no accompanying respiratory, gastrointestinal or urinary symptoms. He was born normally at term. The birth weight was 3.2kg and he had been well prior to this illness. His immunization status was up to date. He was formula fed and was not receiving any supplements including vitamin syrup or fish oil. There was no history of recent travel or of being given unpasteurized milk and there was no major medical or social problems in his family.
On examination, the baby was alert and interestingly, had an “anxious look”. He was irritable, butwas not dysmorphic and looked well nourished. He weighed 6.4kg and his head circumference was41.4cm (both measurements around the 50th centile). He had low grade fever but otherwise, the vital signs were normal. There were no pallor, icterus, lymph node enlargement, hepatosplenomegaly or rashes. He had a mild diffuse fullness on the right side of the jaw which the mother had not noticed earlier. The overlying skin was normal and the swelling was firm in consistency but was mildly tender. The remaining systemic examinations were normal.
Over the next few days, the swelling became more prominent and a similar swelling appeared on the left side of his face. Another swelling of similar nature appeared over the right lower chest a week later. The patient remained afebrile after hospitalization.
Investigations showed microcytic hypochromic anaemia, thrombocytosis and neutrophil count at the upper end of the normal range.
Hb was 9.11g/dl, MCV was 66fl, RDW was 20.1, WBC was 18x109/L, Neutrophil count was 8.1x 109/L, Lymphocyte count was 7.8x 109/L, Platelet count was 887x 109/L and ESR was 105mm/hr. A peripheral blood smear showed hypochromic red cells and thrombocytosis. Sickling was negative. Hence, no other abnormalities were noted.
Serum Amylase was 13U/L(28-100), TP was 72g/L, Alb-was 39g/L, Alkaline phosphatase was
268 U/L, Alanine aminotransferase was 10 U/L, Aspartate aminotransferase was
25U/L, Calcium (total) was 2.61mmol/L, and Phosphate was 2.0mmol/L, while Urea
and Electrolytes were within normal limits. Blood culture revealed no growth and
Maternal Serology for Syphilis (VDRL) was non reactive.
X-rays of Bones showed right mandible and right lower ribs (9 and 10) forming new bone, periosteal reaction and sclerotic changes. However, there were no osteolytic lesions. Urine routine examination was normal. On Ultra sonogram, the abdomen was normal and parotids also looked normal.
The patient was a young infant with irritability and low grade fever who on examination had multiple bony swellings which were slightly tender. Hypervitaminosis was ruled out because he was getting only infant formula and there was no excessive ingestion of the vitamin in any form. A parotitis was considered in the early stages and was excluded by the normal amylase values, ultra sonogram and the subsequent clinical progress. A bone infection was thought to be unlikely because of the lack of “toxicity” with normal vital signs other than a low grade temperature in a “thriving baby” despite multiple sites of involvement. The absence of significant bone tenderness and the radiological picture were against a diagnosis of multifocal osteomyelitis. Congenital Syphilis was very unlikely with a negative serology for syphilis in the mother. A bone tumour again was felt to be very unlikely because of the multiple sites of involvement (primary bone tumours are usually single) and the absence of lytic lesions on X-rays seen in most secondary bone tumours. Scurvy usually does not affect such small infants. The patient had an elevated ESR, persistently high platelet count and the x-rays showed increased bone formation and sclerosis but no lytic lesions. A diagnosis of Caffey disease, although rare, was made in this small infant based on the clinical profile and investigations.
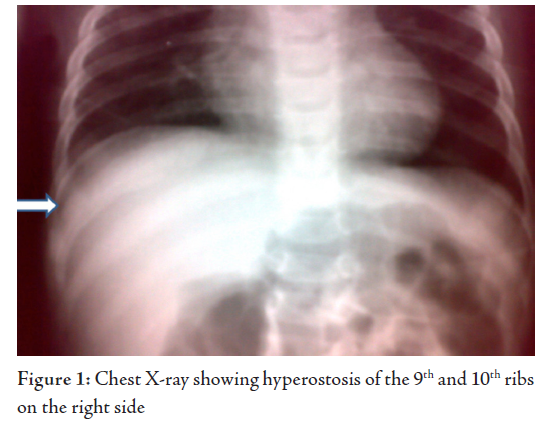
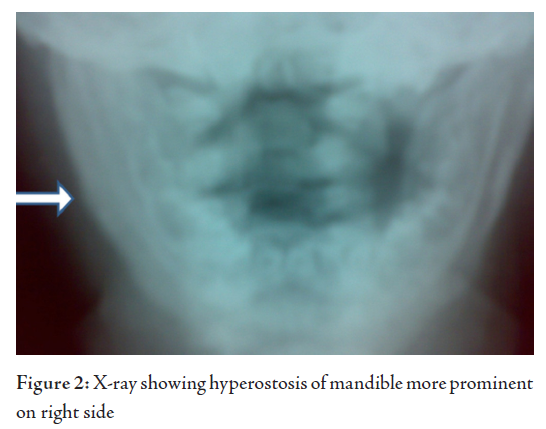
The patient was commenced on Ibuprofen at a dose of 50mg three times a day, and the dose was reduced to twice daily after 3 weeks and was discontinued at week 6. The child became asymptomatic with the jaw and rib swellings significantly reduced in 6 weeks. A review 3 months later showed a healthy child with hardly any swelling in the previously involved sites. X-rays showed good resolution of the hyperostosis. Subsequent review 6 months later showed a normal growing child.
DISCUSSION
Caffey disease, also known as Infantile Cortical Hyperostosis is a self limiting disorder. It is characterized by a triad of systemic symptoms (irritability and fever), soft tissue swelling and underlying cortical bone thickening. It was first reported as a disease entity by Caffey and Silverman in 1945.1 The exact aetiology of this condition is still unknown.2 Most cases are sporadic, but a few familial cases with autosomal dominant and recessive patterns have been described.3 Among the proposed causes are, infections, immunological defects and genetic abnormalities. The discovery of a gene locus in 3 unrelated families with autosomal dominant inheritance (gene COLIAL, 17q21) which encodes Alfa-1 chain of Type I collagen, has raised some doubts whether some cases are a type of Collagenopathy, like Osteogenesis imperfect.4, 5 Similar conditions have also been reported following prolonged treatment with Prostaglandin E1 for maintaining ductal patency in infants with cyanotic heart disease.6,7
The existence of two forms of Caffey disease has been suggested, a classical mild infantile form (ICH) delineated by Caffey and Silverman and a severe form with prenatal onset.8
The condition has been described as rare with no sex or racial predilection. The incidence of the disease appears to fluctuate and other environmental effects may exert an influence.9 Although the exact incidence of the more common classic form of ICH is unknown, there is a world wide decrease in the number of cases particularly the non-familial form. A total of 44 cases have been reported with the severe prenatal onset of Cortical Hyperostosis.2,8,10,11
The classic form has an onset within the first 6 months of life. The manifestations include irritability, swelling of the overlying soft tissue that precedes the cortical thickening of the underlying bones, fever and anorexia. The swelling is painful with a wood like induration but with no redness or warmth, thus suppuration is absent. Mandible is the most commonly involved site followed by scapula, clavicle, ribs and long bones. There are usually no other signs and symptoms. Isolated cases of facial nerve palsy and Erb’s palsy have been reported in the literature.12;13 The pain can be severe and can also result in pseudo paralysis. Other rare clinical findings include dysphagia, nasal obstruction and proptosis.9,14,15 The study patient had none of these uncommon features. Laboratory findings include elevated ESR, and in some patients high alkaline phosphatase, thrombocytosis, anaemia and raised immunoglobulin levels.16,17 The studied patient had elevated ESR, thrombocytosis and anaemia but the alkaline phosphatase was not elevated. Serum immunoglobulin test was not performed.
The severe prenatal onset form is characterized by extensive hyperostotic bone involvement, angulations and shortness of long bones, as well as polyhdramnios and fetal hydrops, which may lead to the incorrect diagnosis of lethal form of Osteogenesis imperfect.8 But absence of other signs like a blue sclera, delicate skin and total absence of fractures and typical histopathological features differentiate this condition from Osteogenesis imperfect.8
Radiography is the most valuable diagnostic study in ICH. Cortical new bone formation (Cortical Hyperostosis) beneath the regions of soft tissue swelling, which is the characteristic feature. While no laboratory tests are specific for diagnosis of ICH, the important differential diagnosis that are to be excluded are osteomyelitis, chronic hypervitaminosis A, bone tumour, scurvy, child abuse and prolonged PGE1 infusion.6,18,19 Awareness of the existence of this rare condition and its typical clinicoradiological profile will avoid the patient being subjected to unnecessary investigation.
Caffey disease is mostly self-limiting and resolves within six months to one year and may not need any treatment.10 However, Indomethacin or Naproxen could be used in really symptomatic cases.18 Steroids can be administered if there is poor response to Indomethacin. In this case, Ibuprofen was used and the outcome appreared to be satisfactory. In some cases, the bone lesions can recur suddenly at their original sites or at newer sites and can have an unpredictable clinical course with remissions and relapses.4,9 Hence, relapse can happen several years later.
CONCLUSION
The aim of this short report is to highlight this disease entity to avoid unnecessary and invasive investigations. The diagnosis of this disease needs an awareness of this condition along with a high index of suspicion. A good history, clinical examination, basic laboratory studies, and plain radiographs are sufficient enough to confirm a diagnosis of this entity in most cases.
The authors reported no conflict of interest and no funding was received for this work.
-
Caffey J, Silverman W. Infantile cortical hyperostosis, preliminary report of a new syndrome. Am J Roentgenol Rad Therapy 1945; 54:1-16.
-
Restrepo S, Sanchez AM, Palacios E. Infantile Cortical Hyperostosis of the Mandible, Ear Nose Throat Journal 2004; 83:454-455.
-
Bernstein RM, Zaleska DJ. Familial Aspects of Caffey Disease. American Journal of Orthopaedics 1996; 24:777-778.
-
Glorieux FF. Caffey Disease a likely Collagenopathy. J Clin Invest 2005; 115:1142-1144.
-
Caffey Disease a type 1 Collagenopathy, Current GGH – Volume 24- No 1. September 2005.www.GGHjournal.com. Accessed on 05.04.2009.
-
Jao Fernando Lourenco de Almeida, Helio K, Luiz H, Hercowitz, Hello K, Eduardo JT. Cortical Hyperostosis secondary to prolonged use of prostaglandin. Clinics Volume 62; No 3, Sao Paulo 2007 Accessed on 03.04.2009.
-
Nadroo AM, Shivangi S, Garg M, Al-Sowaileen. Prostaglandin induced Infantile Cortical Hyperostosis. J Perinatal Med. 2000; 28: 447-452.
-
Susan S, Rabih C, Comelia T, Katharina L, Stephan M, Sigrid T:Antenatal onset of cortical hyperostosis. Am J Med Genet 2003; 120:547-552.
-
Hall C. Caffey disease.Orphanet encyclopedia, Feb 2005 .http://www.orpha.net accessed on April 3rd 2009.
-
Mohammed ALF. Caffey Silverman Disease: Case Report and Literature Review Kuwait Medical journal 2006; 38(1):49-52.
-
Harris VI, Ramilo J. Caffey’s disease. Am J Roentgenol 1978; 130:335-337.
-
Challapalli, Cunningham DG, Varnado SC. Infantile cortical hyperostosis and facial nerve palsy. Int J Pediatr Otorhinolaryngol 1998; 43:175-178.
-
Holtzman D. Infantile cortical hyperostosis of Scapula presenting as Erb’s palsy. J Pediatr 1972; 81:785-88.
-
Sheppard JJ, Pressman H. Dysphagia in Infantile cortical hyperostosis. Dev Med Child Neurol 1988; 30:108.
-
Faure C, Beyssac JM, Montague JP. Predominant orbital and facial involvement in ICH. Pediatr Radiol 1977; 6:103-106.
-
Temperley IJ, Douglas SJ, Rees JP. Raised immunoglobulin and thrombocytosis in infantile cortical hyperostosis. Archives of Disease in Childhood 1972; 47:982-983.
-
Kumar TS, Scott JX, Mathew LG. Caffey Disease with raised Immunoglobulin and Thrombocytosis. Indian Journal of Paediatrics 2008; 75:181-183.
-
Dutta S, Jain N, Bhattacharya A, Mukhopadhyaya K. Infantile Cortical Hyperostosis. Indian Paediatrics 2002; 39:1057.
-
Almada Rodriguez Hugo D. Non accidental injuries in Children-common pit falls Online ISSN: 0972-8074, published on 2005 July 1, accessed on 4th April 2009.